
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The purpose of this work was to develop self-nanomulsifying drug delivery systems (SNEDDS) in sustained-release pellets of ziprasidone to enhance the oral bioavailability and overcome the food effect of ziprasidone. Preformulation studies including screening of excipients for solubility and pseudo-ternary phase diagrams suggested the suitability of Capmul MCM as oil phase, Labrasol as surfactant, and PEG 400 as co-surfactant for preparation of self-nanoemulsifying formulations. Preliminary composition of the SNEDDS formulations were selected from the pseudo-ternary phase diagrams. The prepared ziprasidone-SNEDDS formulations were characterized for self-emulsification time, effect of pH and robustness to dilution, droplet size analysis and zeta potential. The optimized ziprasidone-SNEDDS were used to prepare ziprasidone-SNEDDS sustained-release pellets via extrusion-spheronization method. The pellets were characterized for SEM, particle size, droplet size distribution and zeta potential. In vitro drug release studies indicated the ziprsidone-SNEDDS sustained-release pellets showed sustained release profiles with 90% released within 10 h. The ziprsidone-SNEDDS sustained-release pellets were administered to fasted and fed beagle dogs and their pharmacokinetics were compared to commercial formulation of Zeldox as a control. Pharmacokinetic studies in beagle dogs showed ziprasidone with prolonged actions and enhanced bioavailability with no food effect was achieved simultaneously in ziprsidone-SNEDDS sustained-release pellets compared with Zeldox in fed state. The results indicated a sustained release with prolonged actions of schizophrenia and bipolar disorder treatment.
Introduction
Ziprasidone (), a dopamine receptor antagonist, is an orally active atypical antipsychotic drug used in the treatment of schizophrenia and bipolar disorder (Harvey & Bowie, Citation2005; Patel & Keck, Citation2006; Greenberg & Citrome, Citation2007). The systemic bioavailability of ziprasidone administered intramuscularly is 100%, or 60%, administered orally with food. The absorption of ziprasidone is increased up to two-fold in the presence of food (Miceli et al., Citation2000; Lincoln et al., Citation2010). Reduced oral absorption of ziprasidone in the fasted state cannot be compensated for simply increasing the prescribed dose (Citrome, Citation2009; Fagiolini et al., Citation2010). Most importantly, patients with schizophrenia often eat a poorer diet (McCreadie et al., Citation1998) and do not necessarily take their medicines as instructed, raising the possibility that patients may not be receiving appropriate medication exposure where drug administration occurs outside institutional contexts. Further, a market research shows around 50% of patients with schizophrenia do not fully comply with treatment, and noncompliance is linked to relapse, re-hospitalization, poor outcome and high economic costs (Perkins, Citation2002).
Figure 1. Chemical structure of ziprasidone.
A good amount of research being currently conducted on ziprasidone including fast dissolving tablets, solid nanocrystalline dispersion, complexion and nanosuspensions (Thombrea et al., Citation2011, Citation2012). Improving dissolution rate and pharmacokinetic in the fasted state of ziprasidone can be obtained using those methods, but those formulation exhibited shorter Tmax values and higher Cmax values compared with Geodon in the fed state. Thus, causing inconvenience to patient and fluctuations of plasma concentration A new dosage form with prolonged actions and enhanced bioavailability and no food effect for ziprasidone is desired.
In order to achieve sustained or controlled release of ziprasidone with enhanced bioavailability and no food effect, conventional matrix or coating-based delivery systems may be proposed, which however are bound to be useless because ziprasidone is poorly water-soluble, the solubility of ziprasidone in water is 0.3 μg/ml. The solubility decreases with increasing pH owing to its pKa (∼6) value (Preskom, Citation2005). Reasonable design of delivery system for ziprasidone should take into account of the advantages of both solubilization technology and sustained or controlled release drug delivery systems.
Self-nanoemulsifying drug delivery systems (SNEDDS) have recently gained wide acceptance due to robust formulation perspectives, practical enhancement of solubility and of oral bioavailability of drugs through lymphatic pathways (Singh et al., Citation2009). Several studies have shown that lipophilic compounds are better absorbed when administrated in self-nanoemulsifying formulations (Chae et al., Citation2005; Zhang et al., Citation2012; Dokania & Joshi, Citation2014). From an absorption point of view, SNEDDS are potent in oral bioavailability enhancement. However, owing to their smaller particle size, nanoemulsions reconstituted from SNEDDS can be digested quickly, thus achieving much quick absorption (Porter et al., Citation2007; Pouton & Porter, Citation2008; Khan et al., Citation2014), which may remain the problem of peak and valley fluctuations of plasma drug levels like the previous studies (Thombrea et al., Citation2011, Citation2012).
Pellets offer several advantages of oral sustained-release drug delivery systems, because they travel within the gastrointestinal tract (GIT) under less variable transit times than other forms (Fukumori, Citation1997). Over the past few years, studies on either self-nanoemulsifying pellets (Setthacheewakul et al., Citation2010) or controlled release pellets (Serratoni et al., Citation2007) have been extensively developed. However, studies on SNEDDS sustained-release pellets are limited.
In this study, in order to enhance the bioavailability and overcome the food effect and the fluctuation of plasma concentration of ziprasidone, we developed the ziprasidone-SNEDDS (ZIP-SNDDS) sustained-release pellets to take advantage of the solubility and adsorption enhancing effect of SNEDDS and release rate-controlling capacity of the sustained-release pellets.
Materials and methods
Materials
Ziprasidone was obtained from Jiangsu Nhwa pharmaceutical co., Ltd. (Xuzhou, China). Hydroxy propyl methyl cellulose (HPMC E15) was from ISP Tech., Ltd. (Shanghai, China). Microcrystalline cellulose (MCC) was obtained from the Shanghai Chinaway Pharmaceutical Tech. Co., Ltd. (Shanghai, China). Olive oil, soybean oil, isopropyl myristate (IPM), ethyl oleate, glycerol, polyethylene glycol 200 (PEG 200), polyethylene glycol 400 (PEG 400), Tween 40, Tween 80 and Span 80 were purchased from the Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Capmul MCM, Capmul GMO, Labrasol and Transcutol were purchased from Dow Chemical Co., Ltd. (Shanghai, China).Unless otherwise stated all other materials were of analytical grade.
Methods
Solubility studies
An excess amount of ziprasidone was added to 5 ml of various oils (olive oil, soybean oil, ethyl oleate, IPM, Capmul MCM and Capmul GMO), surfactants (Tween 40, Tween 80, Span 80 and labrasol), and co-surfactants (transcutol, PEG 200, PEG 400 and glycerol) and mixed by vortexing in order to facilitate proper mixing of ziprasidone with the vehicles. The samples were incubated in an air-bath oscillator at 37 ± 0.5 °C for 48 h until equilibrium. The mixtures were then centrifuged at 5000 rpm for 10 min. The supernatant was removed and diluted by methanol, and then analyzed for ziprasidone content using HPLC as described in the section “In vitro drug release”.
Construction of pseudo-ternary phase diagrams
The excipients of oil, surfactant and co-surfactant, selected from the solubility studies were used to construct the pseudo-ternary phase diagrams employing water titration method. Different ratios of oil to surfactant/co-surfactant mixture (Smix) were selected, ranging between 1:9 and 9:1, to delineate the boundaries of nanoemulsion region. The mixture of Smix and oil phase was titrated with water that was added drop wise under vigorous stirring by magnetic stirrers (IKA, Germany) at 150 rpm. Visual monitoring was done during titration and transparency and easy pour ability marked the end point of titration. The pseudo-ternary phase diagrams were constructed by using the software OriginPro 8.6 (Northampton, MA).
Preparation of ZIP-SNEDDS
Once the self-emulsifying region was identified, the desired component ratios of SNEDDS were selected (). Different ZIP-SNEDDS were prepared by selecting the concentration of oil and Smix from pseudo-ternary phase diagrams. The ZIP-SNEDDS were prepared by simple admixture of drug with oil and Smix using vortex mixer at room temperature.
Table 1. Composition of the liquid ZIP-SNEDDS formulations.
Characterization of ZIP-SNEDDS
Self-emulsification time
The efficiency of self-emulsification of ZIP-SNEDDS was assessed by using a standard Chinese Pharmacopoeia type III dissolution apparatus (Shanghai Huanghai Instrument Co., Shanghai, China). 1 ml of each formulation was added to the medium (900 ml of water with a paddle speed of 50 rpm at 37 ± 0.5 °C) by a dropping pipette and the time required for the disappearance of the SNEDDS was recorded (Balakumar et al., Citation2013). The efficiency of self-emulsification was visually assessed.
Effect of pH and robustness to dilution
Formulations were subjected to 50 -, 100 - and 1000-fold dilution with water, 0.1 M HCl and pH 6.8 PBS. The diluted samples were stored for 24 h and observed for any physical changes, such as coalescence of droplets, precipitation or phase separation (Elnaggar et al., Citation2009). The sample with 100-fold dilution with water was monitored as the pH of the ZIP-SNEDDS by pH meter (Leici, Shanghai, China).
Droplet size analysis and zeta potential
In this study, the effect of various dispersion volume on droplet size was investigated. The selected formulation (1 ml) was diluted to 50-, 100- and 1000-fold with water. The droplet size distribution and zeta potential were determined using dynamic light scattering principle (Malvern Zetasizer, Nano ZS-90, Worcestershire, UK). The values of mean droplet size and zeta potential were recorded.
Preparation of ZIP-SNEDDS sustained-release pellets
Based on the results of characterization of ZIP-SNEDDS, the optimal formulation of ZIP-SNEDDS was obtained, the selected formulation was used to prepare ZIP-SNEDDS sustained-release pellets through extrusion-spheronization according to the reported method (Siddique et al., Citation2010; Wang et al., Citation2010). The formulation was prepared by first adding 200 ml 75% ethanol to 30 g ZIP-SNEDDS. Mixing was performed until a homogeneous solution was obtained. Approximately 70 g of HPMC and MCC mixtures (with different weight ratio) were added to the solution and a soft material was obtained. This material was pelletized using an extrusion-spheronization machine (Guanlian Pharmaceutical Equipment Co., Ltd., Shanghai, China). The wet mass was passed through an extruder at 30 rpm. The extrudates were then placed in a spheronizer fitted with a cross-hatched plate rotated at 1800 rpm for 15 min.
Characterization of ZIP-SNEDDS sustained-release pellets
The morphology of the pellets, including their external surfaces and the cross-sections were investigated by scanning electron microscope (SEM) (S-4100, Hitachi, Tokyo, Japan). The pellet formulations were mounted on the stub. The specimen was then coated in a vacuum (6 pa) with platinum using Hitachi Ion Sputter (E-1030) for 240 s at 15 mA.
The particle size of ZIP-SNEDDS sustained-release pellets were measured through photo correlation spectroscopy using a particle size analyzer (BIC 90 plus, Brookhaven Instruments Corporation, Holtsville, NY) at a fixed angle of 90° at 25 °C.
The droplet size distribution and zeta potential of the ZIP-SNEDDS sustained-release pellets were determined using dynamic light scattering principle (Malvern Zetasizer, Nano ZS-90, Worcestershire, UK). ZIP-SNEDDS sustained-release pellets (1 g) were diluted to 100 folds with water in a volumetric flask and was shaken gently and filtered (0.45 μm, PALL, New York city, NY). The values of mean droplet size and zeta potential were recorded.
In vitro drug release
The release profiles of ZIP-SNEDDS, ZIP-SNEDDS sustained-release pellets and Zeldox (all those formulations contained 20 mg ziprasidone) were studied using Chinese Pharmacopoeia type III Apparatus, paddle method with big vessel (Shanghai Huanghai Instrument Co., Shanghai, China) at the paddle rotation speed of 50 rpm in 900 ml dissolution medium, at 37 ± 0.5 °C. At the specified times, 1 ml aliquot was withdrawn, filtered (0.45 μm, PALL, USA) and analyzed by reversed-phase HPLC method (Agilent 1260, Agilent Technologies, Inc., Santa Clara, CA) using a Phenomenex Luna C18 column (4.6 mm × 250 mm, 5 μm) and a 1.0 ml/min flow rate, a 20 μl injection volume, a 30 °C column temperature, 254 nm UV detection. The diluent was 0.06 mol/l ammonium acetate/methanol (15/95, v/v). 1.0 ml of fresh dissolution media pre-warmed to 37 ± 0.5 °C was replaced into the dissolution medium after each sampling. All the experiments were performed in triplicate.
The similarity of release profiles in different release medium (water, 0.1 M HCl and pH 6.8 PBS) was evaluated by similarity factor (f2) recommended by FDA. The similarity factor as a logarithmic transformation of the sum-squared error of differences between the test preparation and reference preparation was calculated by the following equation (Equation Equation1(1)
(1) ):
(1)
(1)
where, Rt and Tt are the accumulated release rates of the reference preparation and test preparation at the predetermined time points, respectively, and n is the number of the time points. The similarity factor fits the result between 0 and 100. It is 100 when the profiles are identical and the f2 approaches 0 as the dissimilarity increases. Generally, the release profiles are considered to be similar if f2 > 50. The larger the f2 value, the higher is the similarity.
Stability studies
Stable SNEDDS sustained-release pellets can be generally stored for a long time under normal storage conditions. Properties such as content and droplet size after dilution should not be significantly changed. In the current research, the stability of the sustained-release pellets was evaluated for three months. The samples of ZIP-SNEDDS sustained-release pellets were maintained in a stability chamber at 30 ± 2 °C and 65 ± 5% relative humidity (RH). The samples were collected after 0, 1, 2 and 3 months. The appearance, droplet size, zeta potential analysis and drug content were evaluated.
Pharmacokinetic studies in dogs
The in vivo pharmacokinetic study described here was reviewed and approved by the Nanjing Medical University Institutional Animal Care and Use Committee (No. 201310113) and adhered to the “Principles of Laboratory Animal Care”. The commercial formulation of Zeldox and ZIP-SNEDDS sustained-release pellets (both of them containing 20 mg ziprasidone) were both tested under fasted (with free access to water between meals) and fed conditions in a parallel study with 12 laboratory beagle dogs, divided into two groups.
In the fasting studies, dogs that had fasted the previous day were dosed and then immediately given 60 ml of tap water by an oral gavage. In the fed studies, dogs that had fasted for 12 h were fed a diet consisting of 150 g dry dog food (10% fat; total fat 15 g). They were dosed and then immediately given 60 ml of tap water by an oral gavage. After dosing, the dogs were returned to metabolism cages with free access to water. They were fed their normal diet after the 12 h sampling time point.
Whole blood samples ∼5 ml were taken from the jugular vein of the dogs using 5 ml disposable syringes with 20 gauge needles immediately before dosing and at the following time points after dosing: 1, 2, 3, 4, 6, 8, 12, 16 and 24 h. Samples were immediately transferred into sodium heparin tubes for anticoagulant. The samples were centrifuged for 10 min at 4000 rpm. The resultant plasma was poured into a 2 ml cryogenic plastic tubes and stored in a freezer (at −20 °C) until assayed.
The samples were extracted using the following procedure: plasma (1.0 ml) was mixed with 40 μl clozapine methanol solution (0.5 μg/ml) as an internal standard, and 5.0 ml ethyl acetate in a 10 ml plastic centrifuge tube, centrifuged at 4000 rpm for 10 min to precipitate the proteins and the supernatant layer was evaporated in a rotary centrifugal vacuum evaporator. The residue was reconstituted in 100 μl mobile phase and 20 μl of the resulting solution was analyzed by HPLC-UV method with Agilent 1260 HPLC (Agilent, CA). Chromatography was performed on a phenomenex Luna C18 column (250 × 4.6 mm, 5 μm); column temperature 30 °C; mobile phase −0.03 M ammonium acetate/methanol (23/77, v/v); flow rate −1.0 ml/min; injection volume −20 μl; UV detection was performed at λ = 254 nm. The mobile phase is different in the HPLC study of “In vitro drug release”, because under the mobile phase of 0.06 mol/l ammonium acetate/methanol (15/95, v/v), the peak of internal standard and substances in the blank plasma cannot be separated very well with ziprasidone.
Method validation results gave linearity of 10–1000 ng/ml with atypical calibration cure of y = 0.0111x + 0.0017 (R2 = 0.9989). Extraction recovery at 50 ng/ml, 250 ng/ml and 500 ng/ml ranged from 82 to 95%; the accuracies were 94.64, 96.58 and 100.6%, respectively; the inter- and intra-day precisions were all below 10%.
The non-compartmental pharmacokinetic parameters were calculated by WINNONLIN® software version 5.2 (Scientific Consulting Inc., Greenville, NC).
Results and discussion
Solubility studies
The purpose of drug solubility in various excipients used in SNEDDS was to provide a robust self-emulsifying region with large boundary size in the pseudo-ternary phase diagram and to form nano-droplet size to improve in vivo behavior (Kommura et al., Citation2001; Zhang et al., Citation2008). The solubility of ziprasidone in various oils, surfactants and co-surfactants is shown in . Highest solubility (41.2 mg/ml) was observed in the Capmul MCM. Capmul MCM is a medium chain monoglyceride, is selected as oil phase, which promotes water penetration, self dispersibility of lipid formulations and have good solvent capacity for drugs. Further, Capmul MCM have partial aqueous solubility, which may help in easy dispersion of drug in aqueous medium (Chambin et al., 2004; Prasad et al., Citation2003; El-Leithy et al., Citation2013), hence it was selected as oil phase for preparation of ZIP-SNEDDS.
Figure 2. Solubility studies of ziprasidone in various oils, surfactants and co-surfactants. Data are expressed as mean ± SD (n = 3).
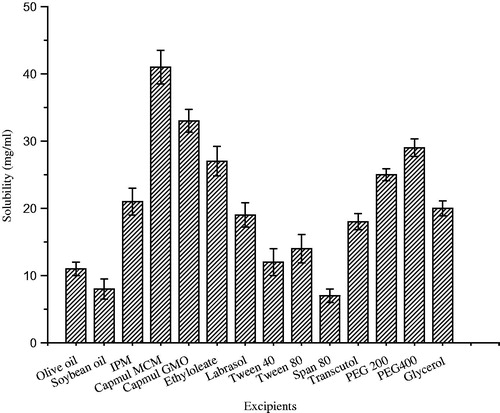
Among the surfactants and co-surfactants, the solubility of ziprasidone was highest in Labrasol (19.1 mg/ml) and PEG 400 (28.8 mg/ml), respectively. Labrasol (polyethoxylated glyceride), a medium chain surfactant, results in interphase curves towards water and favors oil in water emulsion but does not have the capability to decrease the oil–water interfacial tension to the adequate level that can support the formulation of SNEDDS. Thus, the PEG 400 was selected as co-surfactant which helps in further lowering of interfacial tension. Based on the solubility results, the SNEDDS formulation were developed employing Capmul MCM, Labrasol and PEG 400 as oil, surfactant and co-surfactant, respectively.
Construction of pseudo-ternary phase diagrams
Pseudo-ternary phase diagrams were constructed in the absence of ziprasidone to identify the self-emulsifying regions and to optimize the percentage of oil, surfactant and co-surfactant in the SNEDDS formulations. The pseudo-ternary phase diagrams of the systems containing Capmul MCM, Labrasol and PEG 400 as oil phase, surfactant and co-surfactant, respectively, are shown in . There were self-emulsifying region existed with Smix ratios of 3:1, 2:1, 1:1 and 1:2 for Labrasol to PEG 400. It is worth noting that among the tested formulations, the self-emulsifying region is the largest when Smix is 2:1. Thus, in the present study, Smix was determined as 2:1.
Figure 3. Pseudo-ternary phase diagram of systems containing 3:1, 2:1, 1:1, 1:2 Smix ratio Labrasol and PEG 400 using Capmul MCM as oil, and water as titrant.
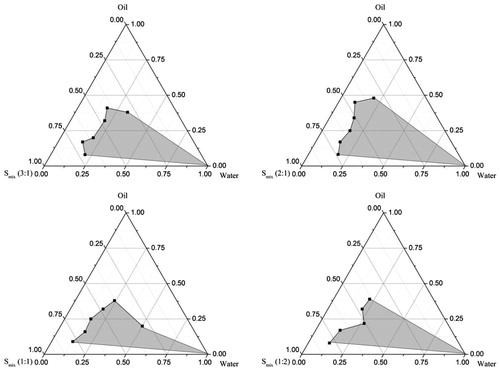
It has been reported that the drug incorporation may have some effect on the self-emulsifying performance (Wei et al., Citation2012). In this study, however, no significant difference was observed in self-emulsifying performance when ziprasidone was added into boundary domain of the pseudo-ternary phase diagram.
Characterization of liquid ZIP-SNEDDS
Self-emulsification time
The rate of emulsification was a major index for assessment of the efficiency of self-emulsification. The SNEDDS should disperse completely and quickly when subjected to dilution under mild agitation (Basalious et al., Citation2010). The emulsification time of formulation F1–F6 were 65 ± 5 s, 77 ± 6 s, 82 ± 6 s, 178 ± 9 s, 221 ± 8 s and 232 ± 7 s. The formulations up to 30% oil content showed the emulsification time of less than 90 s. However, with increase of oil proportion over 40 to 60%, the emulsification time was increased to more than 200 s. Those visual observations indicated that with increased proportion of surfactant/co-surfactant, the greater the spontaneity of emulsification. Considering the side effect of the large quantity of surfactant/co-surfactants, the ratio of Smix was determined as 70%.
Robustness to dilution
Uniform emulsion formation from SNEDDS is very important at different dilutions because drugs may precipitate at higher dilution in vivo, which effects the drug absorption significantly (Constantinides, Citation1995). Different fold dilutions of selected formulation F3 was exposed to different medium to mimic the in vivo conditions where the formulation would encounter gradual dilution. Hence, the formulation was subjected to 50, 100 and 1000 times dilution in water, 0.1 M HCl and pH 6.8 PBS.
The selected formulation was found to be robustness to dilution with different pH conditions. Even after 24 h, formulation F3, showed no signs of precipitation, cloudiness or separation which ensured the stability of the emulsion. The pH of the developed formulation was 6.22 ± 0.05.
Droplet size analysis and zeta potential
Droplet size distribution is one of the most important characteristics of emulsion for stability evaluation and a critical step in the pathway of enhancing drug bioavailability (Xi et al., Citation2009). Droplet size of selected formulation F3 determined was in the range of 54.5 nm to 62.3 nm in different dilution medium with different dilution ratio. The dilution factor had little effect on the droplet size indicating that nanoemulsion was very robust to aqueous volume of the environment.
The charge of the droplets of SNEDDS is another property that should be assessed for increased absorption. Zeta potential of formulation F3 was −28 mV. The high negative charge of the prepared nano-emulsion was probably due to the presence of the free fatty acids. In general, the zeta potential value of ± 30 mV was sufficient for the stability of the system (Muller et al., Citation2001). Hence, the optimized formulation complies with the requirement of the zeta potential for stability.
Preparation and in vitro drug release of ZIP-SNEDDS sustained-release pellets
The selected optimum preparation conditions were as follows: speed of extrusion was 30 rpm, speed of spheronization was 1800 rpm and time of spheronization was 10 min. Four formulations of ZIP-SNEDDS sustained-release pellets () were prepared. These formulations contained a fixed proportion of ZIP-SNEDDS and different ratios of excipients (HPMC and MCC). The in vitro drug release study of the optimized ZIP-SNEDDS, ZIP-SNEDDS sustained-release pellets with different ratios of HPMC and MCC, and Zeldox in water are shown in . Ziprasidone was rapidly released from ZIP-SNEDDS, and an almost complete release was achieved within 30 min, compared with a release rate of less than 25% release from Zeldox within 8 h. The four release profiles of ZIP-SNEDDS sustained-release pellets were different with each other. Increasing the proportion of HPMC may lead to a low-release speed. Therefore, the best sustained release effect can be achieved when the ZIP-SNEDDS:HPMC:MCC ration is 30:40:30 (w/w/w), which had a better sustained released effect than the others.
Figure 4. Cumulative released percent of ziprasidone from ziprasidone-SNEDDS sustained-release pellets with different ratios of HPMC and MCC, ziprasidone-SNEDDS and Zeldox. Data are expressed as mean ± SD (n = 3).
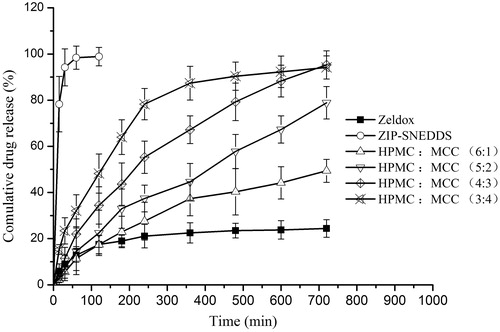
Table 2. Compositions of different ZIP-SNEDDS sustained-release pellet formulations.
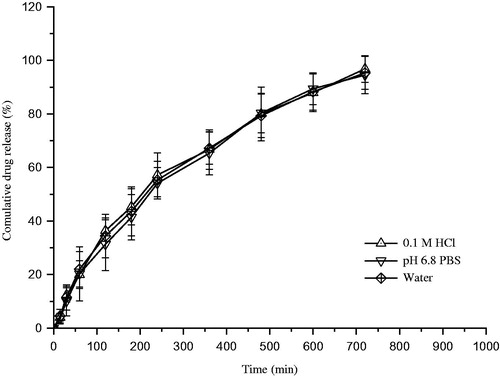
After screening of the ratio of HPMCC to MCC in the formulation, the optimized formulation and drug release profiles in different medium were obtained (). As a result, the influence of medium (0.1 M HCl, pH 6.8 PBS and water) on drug release is not significant. The similarity factor f2 of ziprasidone between water and 0.1 M HCl was 77, the f2 values between water and pH 6.8 PBS was 69. It was indicated that the drug release was independent on the medium pH, which have the potential ability to reduce the effect of pH on the solubility of ziprasidone.
Figure 5. Drug release profiles from ziprasidone-SNEDDS sustained-release pellets with different medium pH. Data are expressed as mean ± SD (n = 3).
Characterization of ZIP-SNEDDS sustained-release pellets
The droplet size of ZIP-SNEDDS sustained-release pellet was 58.4 nm, and zeta potential was 27.7 mV. No obvious difference was found on the droplet size between the ZIP-SNEDDS and ZIP-SNEDDS sustained-release pellets. This result reveals that ZIP-SNEDDS sustained-release pellets can form nano-emulsions and the size is not affected by excipients.
The surface of the pellets and cross-sections were studied by SEM. shows that the pellet had a smooth-surfaced spherical particles. The mean diameter of the pellets was 350 μm.
Figure 6. SEM micrographs of surfaces and cross-sections of ziprasidone-SNEDDS sustained-release pellets. A (150×) and B (2500×) surface of pellets, C (25000×) cross-section of pellets.

Drug content
The drug content in ZIP-SNEDDS sustained-release pellets were conducted as follows: about 200 mg of ZIP-SNEDDS sustained-release pellets were precisely weighed and placed in a 100 ml volumetric flask. The pellets were then diluted with methanol and sonicated for 10 min to extracted ziprasidone completely. The solution was centrifuged at 3000 rpm for 15 min to separate undissolved excipients. The supernatants were filtered through 0.45 μm membrane filter, and detected by HPLC (section “In vitro drug release”). The theoretical content of ziprasidone in the sustained-release pellets was 3% whereas the actual was 2.99%. The ratio of the actual to theoretical content was 99.9%.
Stability studies
The ZIP-SNEDDS sustained-release pellets were kept at 30 ± 2 °C and 65 ± 5% RH, for three months. The results are shown in . The drug content in the SNEDDS sustained-release pellets remained at about 99.85–99.93%, which reflected that the optimized formulation was stable under the experimental condition. Furthermore, no significant change was found in the appearance, ziprasidone content, droplet size and zeta potential analysis.
Table 3. Stability data of ZIP-SNEDDS sustained-release pellets.
Pharmacokinetic studies in dogs
The mean ziprasidone concentration versus time profiles following oral administration of Zeldox (reference) and ZIP-SNEDDS sustained-release pellets to fasted and fed dogs are shown in .
Figure 7. Mean dose-normalized ziprasidone concentration-versus-time profiles after administration of ziprasidone-SNEDDS sustained-release pellets and Zeldox in fasted and fed dogs. Data are expressed as mean ± SD (n = 6).
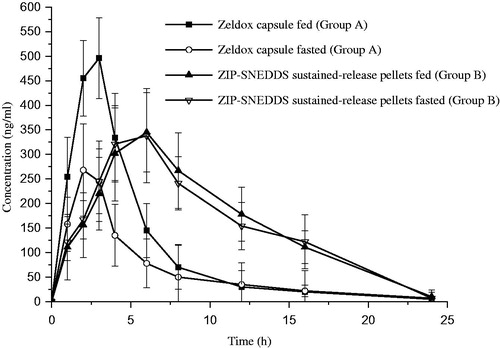
The corresponding mean pharmacokinetic parameters for Zeldox and SD-ZIP ZIP-SNEDDS sustained-release pellets are tabulated in . As the data show, the plasma concentration of ziprasdione from Zeldox capsules increased quickly and reach maximum at 2.5 ± 0.5 h and 1.9 ± 0.9 h in fed and fasted state, and then decreased rapidly. It was evident by the previous reports that the oral Geodone capsules in fed beagle dogs showed the Tmax at 2.3 ± 1.4 h. However, the maximum plasma concentration obtained from ZIP-SNEDDS sustained-release pellets were reached at 6.1 ± 1.0 h and 5.8 ± 1.1 h in fed and fasted state and decreased more slowly than that of Zeldox capsule in the fed state. The Cmax of ZIP-SNEDDS sustained-release pellets were about 345.3 ± 81.5 ng/ml and 338.7 ± 96.5 ng/ml in fed and fasted state, in contrast to 498.4 ± 82.2 ng/ml of Zeldox capsule in the fed state. Meanwhile, the mean resident time (MRT) of ZIP-SNEDDS sustained-release pellets were prolonged significantly to about 11.0 ± 1.5 h and 10.4 ± 2.0 h in fed and fasted state compared with 5.3 ± 1.4 h for Zeldox capsule in the fed state.
Table 4. Pharmacokinetic parameters of ziprasidone in beagle dogs (n = 6) after oral administration of the Zeldox and the ZIP-SNEDDS sustained-release pellets.
The AUC(0–∞) and Cmax for ZIP-SNEDDS sustained-release pellets are similar in the fasted and fed state, indicating essentially no food effect of the drug absorption. Importantly, the Zeldox exhibited a two-fold positive food effect in dogs, which is similar to that obtained with human (Miceli et al., Citation2000). In the present case, a remarkable increase in the AUC of ziprasidone was observed when the SNEDDS pellet was studied. The relative bioavailability of ZIP-SNEDDS sustained-release pellets were 157.8 and 150.1% in fed and fasted state, respectively. The results revealed that the formulation of ziprasidone as an SNEDDS sustained-release pellet significantly promoted and sustained ziprasidone absorption. One unique property of SNEDDS is that it has better absorption in the gastrointestinal tract when loaded with lipophilic drugs (Lu et al., Citation2008). An optimized formulation involving Capmul MCM, Labrasol and PEG 400, which could enhance the solubility of ziprasidone, can also improve absorption. The small droplet size (in the range of 58 nm) of the nanoemulsions may penetrate the absorption site via the transcellular pathway, and could protect the drug from enzyme degradation (Gursoy & Benita, Citation2004). Therefore, the higher bioavailability in ZIP-SNEDDS sustained-release pellets may be due to the enhanced absorption through the lymphatic pathway, as previously reported (Wu et al., Citation2006). A relatively high ratio of emulsifier in SNEDDS may also contribute to the increased permeability by disturbing the cell membrane (Swenson & Curatolo, Citation1992). It was suggested that a controlled release of ziprasidone with prolong actions and enhanced bioavailability and no food effect was achieved simultaneously in ZIP-SNEDDS sustained-release pellets.
Conclusions
In this paper, an improved formulation of ZIP-SNEDDS sustained-release pellet was successfully developed. The in vitro release rate of ziprasidone from the SNEDDS sustained-release pellets exhibited controlled release characteristics with over 95% total release in 12 h. Different dissolution medium had no effect on the release profiles of ZIP-SNEDDS sustained-release pellet. Steady plasma concentration, prolonger Tmax and enhanced bioavailability and no food effect were simultaneously obtained from ZIP-SNEDDS sustained-release pellet, indicating a sustained release with prolonged actions of schizophrenia and bipolar disorder treatment.
Acknowledgements
The authors duly acknowledge the support and scientific inputs provided by Jiangsu Nhwa pharmaceutical co., Ltd. (Xuzhou, China)
Declaration of interest
The authors report no conflicts of interest.
References
- Balakumar K, Raghavan CV, Selvan NT, et al. (2013). Self nanoemulsifying drug delivery system (SNEDDS) of Rosuvastatin calcium: design, formulation, bioavailability and pharmacokinetic evaluation. Colloids Surf B Biointerfaces 112:337–43
- Basalious EB, Shawky N, Badr-Eldin SM. (2010). SNEDDS containing bioenhancers for improvement of dissolution and oral absorption of lacidipine. I: developmentand optimization. Int J Pharm 391:203–11
- Chae GS, Lee JS, Kim SH, et al. (2005). Enhancement of the stability of BCNU using self-emulsifying drug delivery systems (SEDDS) and in vitro antitumor activity of self-emulsified BCNU-loaded PLGA wafer. Int J Pharm 301:6–14
- Chambin O, Jannin V, Champion D, et al. (2004). Pourcelot, influence of cryogenic grinding on properties of a self-emulsifying formulation. Int J Pharm 278:79–89
- Citrome L. (2009). Using oral ziprasidone effectively: the food effect and dose-response. Adv Ther 26:739–48
- Constantinides PP. (1995). Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res 12:1561–72
- Dokania S, Joshi AK. (2014). Self-microemulsifying drug delivery system (SMEDDS)-challenges and road ahead. Drug delivery 1–16
- El-Leithy ES, Ibrahim HK, Sorour RM. (2013). In vitro and in vivo evaluation of indomethacin nanoemulsion as a transdermal delivery system. Drug Deliv 1–8
- Elnaggar YS, El-Massik MA, Abdallah OY. (2009). Self-nanoemulsifying drug delivery systems of tamoxifen citrate: design and optimization. Int J Pharm 380:133–41
- Fagiolini A, Canas F, Gallhofer B, et al. (2010). Strategies for successful clinical management of schizophrenia with ziprasidone. Expert Opin Pharmacother 11:2199–220
- Fukumori Y. (1997). Coating of multiparticulates using polymeric dispersions. In: Ghebre-Sellassie I, ed. Multiparticulate oral drug delivery. New York: Marcel Dekker, 79–111
- Greenberg WM, Citrome, L. (2007). Ziprasidone for schizophrenia and bipolar disorder: a review of the Clinical trials. CNS Drug Rev 13:137–77
- Gursoy RN, Benita S. (2004). Self-microemulsifying drug delivery systems (SMEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother 58:173–82
- Harvey PD, Bowie CR. (2005). Ziprasidone: efficacy, tolerability, and emerging data on wide-ranging effectiveness. Expert Opin Pharmacother 6:337–46
- Khan AW, Kotta S, Ansari SH, et al. (2014). Self-nanoemulsifying drug delivery system (SNEDDS) of the poorly water-soluble grapefruit flavonoid Naringenin: design, characterization, in vitro and in vivo evaluation. Drug Deliv 1–10
- Kommura TR, Gurley B, Khan MA, Reddy IK. (2001). Self-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessment. Int J Pharm 212:233–46
- Lincoln J, Stewart Mark E, Preskorn Sheldon H. (2010). How sequential studies inform drug development: evaluating the effect of food intake on optimal bioavailability of ziprasidone. J Psychiatr Pract 16:103–14
- Lu JL, Wang JC, Zhao SX, et al. (2008). Self-microemulsifying drug delivery system (SMEDDS) improves anticancer effect of oral 9-nitrocamptothecin on human cancer xenografts in nude mice. Eur J Pharm Biopharm 69:899–907
- McCreadie R, Macdonald E, Blacklock C, et al. (1998). Dietary intake of schizophrenic patient in Nithsdale, Scotland: case-control study. BMJ 317:784–5
- Miceli JJ, Wilner KD, Hansen RA, et al. (2000). Single- and multiple-dose pharmacokinetics of ziprasidone under nonfasting conditions in healthy male volunteers. Br J Clin Pharmacol 49:5S–13S
- Muller RH, Jacobs C, Kayser O. (2001). Nanosuspensions as particulate drug formulations in therapy rationale for development and what we can expect for the future. Adv Drug Deliv Rev 47:3–19
- Patel NC, Keck PE. (2006). Ziprasidone: efficacy and safety in patients with bipolar disorder. Expert Rev Neurother 6:1129–38
- Perkins DO. (2002). Predictors of noncompliance in patients with schizophrenia. J Clin Psychiatr 63:1121–8
- Porter CJ, Trevaskis NL, Charman WN. (2007). Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov 6:231–48
- Pouton CW, Porter CJ. (2008). Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev 60:625–37
- Prasad RYV, Puthli SP, Eaimtrakarn S, et al. (2003). Enhanced intestinal absorption of vancomycin with labrasol and D-α-tocopheryl PEG 1000 succinate in rats. Int J Pharm 250:181–90
- Preskom SH. (2005). Pharmacokinetics and therapeutics of acute intramuscular ziprasidone. Clin Pharmacokinet 44:1117–33
- Serratoni M, Newton M, Booth S, Clarke A. (2007). Controlled drug release from pellets containing water-insoluble drugs dissolved in a self-emulsifying system. Eur J Pharm Biopharm 65:94–8
- Setthacheewakul S, Mahattanadul S, Phadoongsombut N, et al. (2010). Development and evaluation of selfmicroemulsifying liquid and pellet formulations of curcumin, and absorption studies in rats. Eur J Pharm Biopharm 76:475–85
- Siddique S, Khanam J, Bigoniya P. (2010). Development of sustained release capsules containing coated matrix granules of metoprolol tartrate. AAPS Pharm SciTechnol 11:1306–14
- Singh B, Bandopadhyay S, Kapil R, et al. (2009). Self-emulsifying drug delivery systems (SEDDS): formulation development, characterization, and applications. Crit Rev Ther Drug Carrier Syst 26:427–521
- Swenson ES, Curatolo WJ. (1992). Means to enhance penetration. Adv Drug Deliv Rev 8:39–42
- Thombrea AG, Herbig SM, Alderman JA. (2011). Improved ziprasidone formulations with enhanced bioavailability in the fasted state and a reduced food effect. Pharm Res 28:3159–70
- Thombrea AG, Shah JC, Sagawa K, Caldwell WB. (2012). In vitro and in vivo characterization of amorphous, nanocrystalline, and crystalline ziprasidone formulations. Int J Pharm 428:8–17
- Wang ZY, Sun J, Wang YJ, et al. (2010). Solid self-microemulsifying nitrendipine pellets: preparation and in vitro/in vivo evaluation. Int J Pharm 383:1–6
- Wei Y, Ye X, Shang X, Peng X. (2012). Enhanced oral bioavailability of silybin by a supersaturatable self-emulsifying drug delivery system (S-SEDDS). Colloid Surf A: Physicochem Eng Aspects 396:22–8
- Wu W, Wang Y, Que L. (2006). Enhanced bioavailability of silymarin by selfmicroemulsifying drug delivery system. Eur J Pharm Biopharm 63:288–94
- Xi J, Chang Q, Chan CK, et al. (2009). Formulation development and bioavailability evaluation of a self-nanoemulsified drug delivery system of oleanolic acid. AAPS Pharm Sci Tech 10:172–82
- Zhang P, Liu Y, Feng N, Xu J. (2008). Preparation and evaluation of self microemulsifiying drug delivery system of oridonin. Int J Pharm 355:269–76
- Zhang Y, Wang R, Wu J, Shen Q. (2012). Characterization and evaluation of self-microemulsifying sustained-release pellet formulation of puerarin for oral delivery. Int J Pharm 427:337–44