
Abstract
The aim of this study was to obtain a stable, amorphous solid dispersion (SD) with Soluplus, prepared by hot-melt extrusion (HME) as an effective and stable oral delivery system to improve the physical stability and bioavailability of the poorly water-soluble simvastatin (SIM), a drug with relatively low Tg. The drug was proved to be miscible with Soluplus by calculation and measurements. The solubility, dissolution, thermal characteristics, interactions and physical stability of the SIM/Soluplus SDs were investigated. The crystal state of simvastatin in the SD was found to change from crystalline to amorphous form during the HME process and also hydrogen bonds were observed between SIM and the extruded Soluplus. The phase solubility showed the solubilization effect of Soluplus was strong and spontaneous. The equilibrium solubility illustrated that Soluplus/SIM SDs gained much higher solubility than its corresponding physical mixtures (PMs). Both of the dissolution profiles and in-vivo performance showed that the SIM/Soluplus SD obtained a marked enhancement, compared with the PM. There was a little change in the SIM/Soluplus SD during a 3-month storage period (40 °C, 75%), indicating the good physicochemical stability. The extruded Soluplus system prepared by HME is a good alternative for the water-insoluble SIM to improve the stability and bioavailability.
Introduction
Many of these new chemical entities (NCEs) failed to be used in clinical therapy as they have poor bioavailability due to their limited solubility or slow dissolution rate in the gastrointestinal tract (Hong et al., Citation2014). Converting crystalline drugs into their amorphous counterparts is an efficient way to improve the dissolution rate and solubility of poorly water-soluble drugs, especially for biopharmaceutics classification system (BCS) Class II compounds (Srinarong, Citation2011).
A variety of methods have been used to obtain amorphous drugs, such as hot-melt extrusion (HME), spray drying, solvent co-precipitation and co-milling. With the virtue of all these processes, the crystalline drugs can convert to the amorphism, with high internal energy, and this lead to enhanced dissolution and bioavailability. But the amorphous state is easy to decompose and also tend to go back to the natural crystalline one, which is called physicochemical instability (Ambike, Citation2005).
HME technique is well-known to be an efficient way to enhance the dissolution (Srinarong et al., Citation2011). It is a continuous, simple and efficient process not requiring a solvent, aimed at preparing a molecular dispersion or glass solution of the drug in a matrix (Dexia, Citation2013). Also, throughout the 1-min extrusion process, the mechanical energy used and the short heating time will not produce any significant decomposition in the somewhat thermolabile drug (Jijun, Citation2011). However, the study found that a drug, with low Tg value, was hard to obtain stable solid dispersion.
Simvastatin (), one of the most popular cholesterol-lowering drugs, is a class II drug in the Biopharmaceutics Classification Scheme and thus has satisfactory permeability but poor solubility. With a relatively low-glass transition temperature value, it was chosen as a model drug in the study. On the one hand, SIM was reported to decompose easily (Celestino, Citation2012). On the other hand, as stated earlier, the amorphous state is not physicochemically stable compared with its crystalline counterpart (Ahlneck & Waltersson, Citation1986). Therefore, it is significant to choose a suitable formulation and method to get relatively stable amorphous drug-polymer dispersions with enhanced bioavailability (Telang et al., Citation2009).
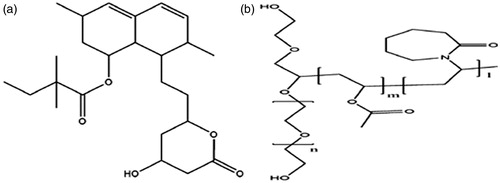
Figure 1. Structure of simvastatin (a) and Soluplus (b).
HME process is always applied in combination with hydrophilic or amphiphilic polymers to improve the dissolution. In the previous studies, PEG, HPMC and PVP have been chosen as carriers to prepare SIM solid dispersions. Silva (Citation2010) found that SIM SDs with PEG 6000 or PVA K15 enhanced the bioavailability significantly, while Pandya (Citation2008) reported that solid dispersions prepared with PEG and HPMC improved the bioavailability without any crystalline change. And as an important part of the formulation, the properties of the polymer function a lot in the stability of the SD.
Soluplus (), an amphiphilic co-polymer, composed of PEG, polyvinyl caprolactam and polyvinylacetate was used in this study. With a Tg of 70.5 °C, Soluplus had a wide extrusion temperature range (Zhang, Citation2013). Also its high flow-ability and excellent extrudability made it suitable for the extrusion process (Hong et al., Citation2014). Numerous studies based on Soluplus have shown that it can enhance the dissolution rates of poorly soluble drugs. Kalivoda et al. (Citation2012) have reported that an extrudate with Soluplus produced a huge increase in dissolution in comparison with that of commercial capsules. Linn et al. (Citation2012) found that Soluplus increased the intestinal absorption of three BCS II drugs, danazol, fenofibrate and itraconazole. Not only can it enhance the solubility and dissolution of poorly water soluble drugs, but it also stabilizes solid dispersions. In fact, solid dispersions of itraconazole/Soluplus prepared by Zhang et al. (2013) markedly enhanced bioavailability and stability. Djuris (Citation2013) discovered that SD prepared with Soluplus increased the in vitro dissolution of carbamazepine and maintained its physicochemical stability. And Kalivoda et al. (Citation2012) also found that a solid dispersion containing Soluplus exhibited the greatest stability.
The object of the study was to develop solid dispersion with Soluplus as an effective and stable oral delivery system for poorly water-soluble SIM to enhance the dissolution and bioavailability. In the study, the solid dispersion was characterized by differential scanning calorimetry (DSC), powder X-ray diffraction (PXRD) and Fourier transform infrared spectroscopy (FT-IR) to monitor its state and interaction. Both the in-vivo and in-vitro performance of the SIM/Soluplus SD and SIM were examined. In addition, SIM/Soluplus SD which had been stored for 3 months (40 °C, RH 75%) underwent an examination involving DSC, dissolution testing and HPLC, in order to monitor its stability.
Materials and methods
Materials
SIM was purchased from Zhengzhou Ruikang Pharmaceutical Company (Zhengzhou, Henan, China). Talc was provided by Guangxi Huashi Chemical Company (Guangxi, China) and BHA was obtained from Guangxi Huashi Chemical Company (Guangxi, China), while PVPP was purchased from International Specialty Products (Beijing, China). MCC was obtained from Asahi Kasei (Tokyo, Japan) and Soluplus was kindly provided by BASF (Shanghai, China). Commercial tablets (Zocor®, SIM 20 mg), used as a reference, were purchased from MSD (Hangzhou, China).
Samples preparation
Preparation of SIM/Soluplus physical mixtures
Formulations containing 20, 40, 60, 70, 80, 90% Soluplus and 0.1% BHA were accurately weighed and mixed by hand till a physical mixture was obtained. PMs were then passed through a 100-mesh screen (Chokshi, Citation2007), and stored in a desiccator until required.
Preparation of SIM/Soluplus SD by HME
A COPERION KEYATE-20 (Nanjing, China) twin-screw extruder was used to prepare the SIM/Soluplus SD. The temperatures of the extruder barrel zones and die were controlled as follows: Zone 1 = 150 °C, Zone 2 = 150 °C, Zone 3 = 150 °C, Zone 4 = 150 °C and Die = 80 °C. The feed rate and screw rate were both 2.5 Hz. PMs were simultaneously melted, homogenized and extruded in the chamber. Then the extrudates were collected and cooled at room temperature, then grounded. Finally, the SDs were passed through a 100-mesh sieve, and stored in a desiccator until required (Sun, Citation2008; Dexia, Citation2013).
Preparation of SIM/Soluplus SD–T via direct compression method
The SIM/Soluplus SD containing 80% Soluplus was mixed by hand for 5 min with MCC (41.9%), PVPP (3%) and talc (5%) to obtain the formulation blend. And it was passed through sieve no.60. Then the content of SIM was determined to calculate the weight. Finally, the mixture was compressed into convex faced tablets weighing 200 mg, with 5 mm diameter by a single punch tablet machine. The compression force and time was 20 kN and 32 s, respectively (Surampalli & Nanjwade, Citation2014).
Characterization of SIM/Soluplus SD
Hansen solubility parameter calculations
The Hansen solubility parameters of the drug and the polymers were calculated from their chemical structures using the Hoftyzer and Krevelen method (Mididoddi & Repka, Citation2007) according to the following equation:
The total solubility parameter (δt) is determined from the interactions between dispersion forces (δd), hydrogen bonds (δh), and polar interactions (δp) of the functional groups in the parent molecule. For polymeric excipients, determination of the solubility parameter was based on the average molecular weight.
Differential scanning calorimetry
Differential scanning calorimetry (DSC) was performed using a differential scanning calorimeter-60 and Thermal Analyzer-60 WS (Shimadzu, Kyoto, Japan). Samples weighing 3–5 mg were spread out in hermetic aluminum pans under a dry nitrogen purge, and analyzed at a heating rate of 10 °C/min from 30 °C to 150 °C.
X-ray powder diffraction
A D/Max-2400 X-ray Fluorescence Spectrometer (Rigaku, Tokyo, Japan) with Cu-Kα radiation was used to carry out X-ray powder diffraction (XRPD) measurements. The equipment was set to a voltage of 56 kV, a current of 182 mA and a scanning rate of 0.2°/min over a 2θ range of 5°–60°.
Scanning electron microscopy
Samples were analyzed with a scanning electron microscope (SSX-500, Shimadzu, Japan) operated at an acceleration voltage of 15 kV.
High-performance liquid chromatography
To determine the degradation, the content of the samples was examined by high-performance liquid chromatography (HPLC) using methods described in the CP. A Hitachi series 2100 liquid chromatograph (Hitachi, Tokyo, Japan) consisting of a PU-2130 pump, an autosampler and a UV-2400 UV detector were used for determination. Chromatographic separation was achieved using a Venusil ASB C18 analytical column (4.6 mm × 25 mm, particle size 3 μm).
Interactions between the amorphous SIM and Soluplus
Prediction the interactions by the Tg values
The Tg value was determined by the method reported by Zhang (Citation2009). The above samples remained at 150 °C for 2 min in order to remove any residual moisture, then quenched at −20 °C/with a cooling rate of 200 °C/min and kept at that temperature for 3 min, then scanned again up to 100 °C at a rate of 10 °C/min. The middle point of the data was recorded as the Tg value. All measurements were carried out in triplicate.
In addition, the Tg value can also be estimated. In binary systems, the Tg value of the amorphous one-phase dispersion is calculated using the Fox equation (Tajber et al., Citation2005):
where W1 and W2 are the weight fractions of SIM and Soluplus, respectively, and Tg1 and Tg2 are the corresponding glass transition temperatures.
Confirmation of the existing interactions
FT-IR was conducted on a BRUKER IFS 55 FT-IR system using the KBr disk method to confirm the existing interactions. The spectra were recorded at a resolution of 4 cm−1 over the range 4000–400 cm−1.
Solubility study
Determination of phase solubility
Solubility measurements were performed in triplicate. An excess of SIM was added to Soluplus solutions of increasing concentrations from 0 to 30% and then they were sealed and shaken at 25 ± 0.5 °C for 48 h in a thermostatically controlled water bath. Samples were then withdrawn and passed through a 0.22 μm membrane filter, and finally determined by HPLC.
The Gibbs free energy of SIM for the transfer of pure water to aqueous solutions of Soluplus was calculated using the following equation (Kadam, Citation2011):
where Sc/So is the ratio of the molar solubility of SIM in aqueous solution of Soluplus to that in pure water, R is the general gas constant and T is the absolute temperature. The value of the apparent stability constant, Ks (Ahuja et al., Citation2007), between the drug–carrier combinations was calculated from the phase solubility profiles, as described below:
Determination of equilibrium solubility
An excess of SIM, PM, SD was added to distilled water, and then it was sealed and shaken at 25 ± 0.5 °C for 48 h in a thermostatically controlled water bath. Then, the solution was withdrawn, passed through a 0.22 μm membrane filter, and finally determined by HPLC.
Dissolution test
Each sample for the dissolution test contained 20 mg SIM. The medium was 900 ml of 0.01 M sodium dihydrogen orthophosphate solution containing 0.2% w/v sodium dodecyl sulphate with a pH of 7.0 (adjusted using 50% sodium hydroxide). The rotation speed of the paddles was 50 r/min. Samples (6 ml) were withdrawn from the medium at intervals (5, 10, 20, 30, 45, 60 min), then filtered and subjected to HPLC with detection at 238 nm.
Bioavailability study
The study protocol was approved by the Ethics Committee of Shenyang Pharmaceutical University. Bioavailability studies were performed using six beagle dogs, which were fasted overnight (12 h) before oral administration. The dogs were divided into two groups and given SIM-SD-Ts and commercial tablets (containing 20 mg SIM), respectively. The experiment was conducted according to the method reported in the literature [20]. Blood samples (0.5 ml) were taken after 0.17, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12 and 24 h by retro-orbital puncture and transferred to heparinized tubes. Plasma was separated from the blood samples by centrifugation at 4000 rpm for 10 min and then stored at −20 °C until required for analysis. Determination of simvastatin in plasma samples was carried out using an ACQUITYTM UPLC system (Waters Corp., Milford, MA) with a cooling autosampler and column oven as reported in the literature (Luo, 2013). The separation method involved gradient elution using a mobile phase composed of 0.01 M ammonium acetate water and acetonitrile. A Waters ACQUITYTM TQD triple quadrupole tandem mass spectrometer (Waters Corp., Manchester, UK) with an electrospray ionization (ESI) interface was used for mass analysis. The method validation results were as follows: the RSD values reflecting intra-day and the inter-day precision were both not more than 10.0%; the extraction recoveries of SIM from the three different QC plasma samples were 72.2, 79.8 and 72.0%; the extraction recoveries of SIM from the three different QC plasma samples were 54.2, 52.3 and 51.4%; and the mean extraction recovery of LOV was 73.3%.
Preliminary stability test
SIM SD was sealed tightly with aluminum-plastic packing and tested during storage (40 °C, RH 75%). The stability was evaluated in three different ways: DSC, dissolution testing and HPLC. The methods were described above in detail. SIM SD was tested after storage for 1, 2 and 3 months.
Results and discussion
Miscibility of SIM and Soluplus
The solubility parameters of drug and polymer were calculated to investigate the miscibility of SIM and Soluplus. Greenhalgh et al. have demonstrated that compounds with an Ää2t <7.0 Mpa1/2 are likely to be miscible but when the Ää2t >10 Mpa1/2, the compounds are immiscible. While, if the value of Ää2t is in the range 7.0–10 Mpa1/2, the compounds can be partly miscible (Avachata et al., Citation2012). From , the calculated value of Ää2t was 2.8 MPa1/2, which was significantly below 7.0 MPa1/2. The small difference between the calculated solubility parameters of Soluplus and SIM showed that SIM was likely to be miscible with Soluplus. This result is in accordance with the DSC analysis as shown in . As is shown, there was a typically sharp endothermic peak of the pure SIM at 139 °C, which became lower and broader in the PMs, along with a shift in temperature to about 126 °C. This phenomenon indicated that SIM could gradually dissolve in Soluplus during the HME process (Ahlneck & Waltersson, Citation1986). In addition, the Tg value of the SIM/Soluplus SD () was single, supporting the conclusion above. Therefore, Soluplus is a good alternative for the oral delivery system.
Figure 2. DSC thermograms of (a) SIM; (b) Soluplus; (c) 80% SIM/Soluplus SD; (d) 80% SIM/Soluplus PM.
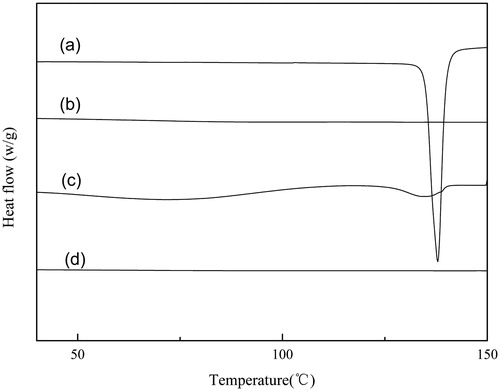
Table 1. Calculated solubility parameters of drug and polymers.
Table 2. Tg values of freshly prepared SIM/Soluplus solid dispersions (by the second heating cycle).
Characterization of solid dispersion
Differential scanning calorimetry
As shown in , simvastatin is a crystalline drug, with a sharp melting point Tm at 139.5 °C, while Soluplus is amorphous without a melting endothermic peak. The absence of the typical peak in SIM/Soluplus SD indicated that simvastatin was amorphous in the extruded Soluplus, implying that the morphology of SIM changed during the hot melt extrusion process (Hu et al., Citation2006; Sarode, Citation2013). However, in the corresponding PM, the endothermic peak was still observed, though it became much weaker.
Powder X-ray diffraction
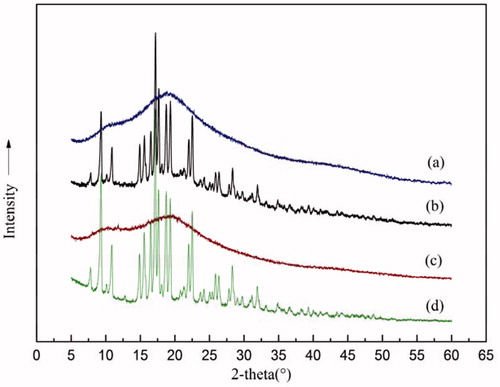
Powder X-ray diffraction (PXRD) was performed and results are presented in . As can be seen, the SIM crystals displayed clear diffraction peaks in the range 8–32° (Telang et al., Citation2009)], and in that range, the amorphous Soluplus showed no diffraction peaks at all. The absence of the diffraction peaks in SIM/Soluplus SD indicated that simvastatin was amorphous in SD, conforming to the conclusion that the morphological change occurred during the HME process (stated in the above sub-section).
Figure 3. PXRD patterns of (a) 80% SIM/Soluplus SD; (b) 80% SIM/Soluplus PM; (c) Soluplus; (d) SIM.
Scanning electron microscopy
The microphotographs of pure SIM and its SD are shown in . The morphology of pure drug () was crystalline, while in the SD, no crystal lattice was observed, revealing that significant changes occurred in the shape and surface topography during the HME process. The analysis confirmed the conclusion that there was a crystal transition of SIM from the crystalline to the amorphous state in the SD (Zhang, Citation2013).
Figure 4. FTIR spectra of 80% SIM/Soluplus SD and the corresponding PM; possible forms of hydrogen bonds between Soluplus and SIM.
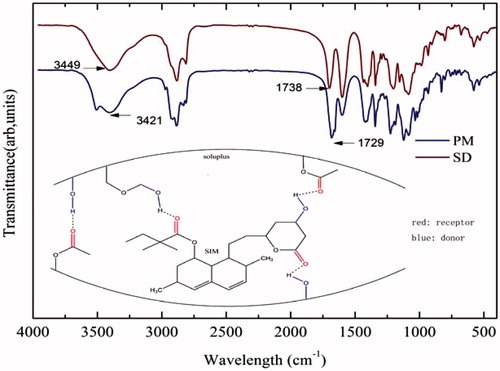
Figure 5. SEM images of (a) SIM crystalline; (b) ITZ/Soluplus (20:80) solid dispersion prepared by HME obtained by scanning electron microscopy.

Interactions between the amorphous SIM and Soluplus
Prediction of the intermolecular interactions
Hydrophilic carriers are known to interact with drug molecules mainly by van der Wales forces and hydrogen bonds.
DSC analyses of the glass transition temperature of SDs in different drug/polymer ratios are shown in , where the estimated Tg values were compared with the measured ones. In general, both of the values decreased as the content of Soluplus reduced. This is because SIM has a Tg value at 301.13 K, which is close to the 298.5 K reported in the literature (Zhang, Citation2009) and much lower than that of Soluplus at 343.5 K. In the system, Soluplus acted as an anti-plasticizer, with the ability to effectively increase the Tg (Sousa, Citation2010).
Figure 6. Tg values of freshly prepared SIM/Soluplus solid dispersions (by the second heating cycle) and the ones predicted by the Fox equation.
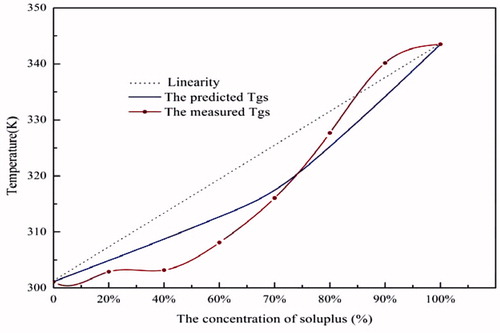
The Fox equation was applied to predict the Tg values (). And there were deviations between the estimated and the measured ones. The negative deviation illustrated that the interactions between the drug and polymer were not strong and it occurred mainly between like molecules and might lead to phase separation, while, if the deviation was positive, this indicated that the drug was miscible with polymer and the interactions contributed to stabilizing the system (Tajber et al., Citation2005; Zhang, Citation2009; Barmpalexis, Citation2013).
can be divided into two sections. When the concentration of Soluplus was over 80%, the calculated Tg value was greater than the measured ones, indicating the presence of interactions between SIM and Soluplus. While, when the Soluplus concentrations fell below 80%, the measured ones from DSC exhibited a negative deviation from the predicted values, indicating that the interactions between SIM and Soluplus were weaker than the like molecules.
Confirmation of the existing interactions
As shown in the section “Differential scanning calorimetry”, there were interactions between the drug and carrier. FT-IR () was conducted to examine the interactions in detail.
From the chemical structure of SIM and Soluplus (), it can be assumed that interactions occurred between the lipophilic portions of Soluplus and SIM. The analysis of the spectra was then focused on the regions between 16 significant peak shifts following hydrogen bonding could be detected there (Jun, Citation2007; Zhang, Citation2009).
The IR spectrum of SD should have been an integration of SIM and Soluplus. However, the ratio of SIM to Soluplus was very low and the characteristic peaks of SIM were covered by those of Soluplus. Also the conversion of SIM from a crystalline form to an amorphic one in the solid dispersion that made the peaks of SIM become weaker and broader, and even completely covered by the peaks of Soluplus. Then, the shift in the spectrum, mainly involved the functional groups of Soluplus. The -OH region in SD turned out to appear at 3421 cm−1 (the wavelength of the corresponding PM was 3449 cm−1) with a weaker and broader peak. Also in the stretching vibration of the ester carbonyl functional group, there was a shift from 1738 cm−1 to 1729 cm−1. These shifts indicate the presence of intermolecular hydrogen bonding (in ), which was in agreement with previous results for amorphous SIM (Lobmann, Citation2012).
In vitro performance
Solubility study
The solubility of the drug in the presence of concentrated solutions of a polymeric carrier can be used to analyze the mechanism of dissolution from an SD. There are many reports in the literature that report Soluplus as a solubilizer (Shamma & Basha, Citation2013). The equilibrium solubility of SIM in Soluplus solution of different concentrations was tested to see the solubilization effect of Soluplus. shows that the solubility of SIM linearly increased with the Soluplus levels, in accordance with the results of a previous study (Pandya, Citation2008; Shamma & Basha, Citation2013).
Figure 7. Phase solubility of SIM in Soluplus solutions with different concentrations. (25 °C, pure water, n = 3).
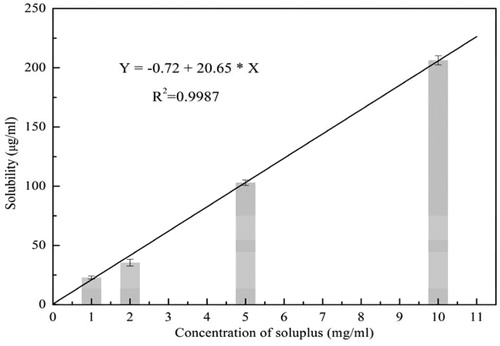
Furthermore, the Gibbs free energy was used to examine the process of transfer of SIM from pure water to aqueous Soluplus solution. The values of are presented in and all were negative, indicating the spontaneous nature of the solubilization (Kadam, Citation2011). Also the values declined with an increase in the polymer concentrations, further illustrating the solubilization effect of Soluplus.
Table 3. Thermodynamic parameters of SIM in Soluplus solutions with different concentrations.
Soluplus, a hydrophilic carrier, interacts with drug molecules mainly by hydrogen bonding (as stated in the section “Interactions between the amorphous SIM and Soluplus”). The values of the apparent stability constant Ks were calculated to examine the strength of these interactions. The Ks value of the AL-type system was less than unity, which indicated that interactions occurred (Ahuja et al., Citation2007). Also, the high value of Ks relative to that reported in the literature showed that the binding affinity between SIM and the solubilizer was strong.
In addition, the solubility of SD with 80% Soluplus in pure water displayed a significant increase to about 52.13 μg/ml, while that of plain drug was 0.51 μg/ml, exhibiting an increase of over 100-fold. Also, for the corresponding physical mixture, the solubility of SD exhibited a 2-fold increase.
The improvement in solubility of SD was mainly due to the conversion to high energy, amorphous dispersions in amphiphilic polymers, which helped SIM obtain a higher wettability, undergo favorable drug-polymer interactions (Hong et al., Citation2014). Also, a previous report showed that the drug in Soluplus achieved micelle solubilization, which markedly improved the drug solubility (Yu, Citation2013). In fact, in the phase solubility test, emulsified opalescence was indeed observed in the Soluplus solution.
The dissolution test
The dissolution of SD-T and the commercial tablet are displayed in . The SD tablet needed more time to achieve the highest release compared with the corresponding SD, which may be attributed to the slower disintegration of the tablet (Barmpalexis, Citation2013). The SD-T achieved the highest release at 96.4% in 20 min, which was about 2-fold higher than that of the commercial tablet. And compared with the raw drug and the corresponding PM, the SD achieved almost 3.8- and 2-fold higher dissolution, respectively.
Figure 8. Dissolution profiles of different formulations. Each vessel (n = 3) contained the equivalent of 20 mg simvastatin and 900 ml 0.1% SDS solution using CP apparatus II with paddles at 50 rpm.
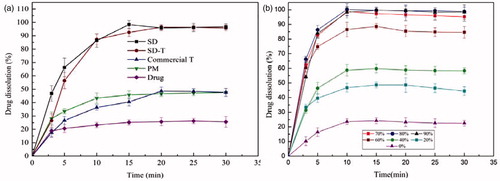
The dissolution profiles of SDs with different Soluplus/drug ratios are also shown in . The drug dissolution of SDs with 70–90% Soluplus was much higher than that of SDs containing <70% Soluplus. The order of dissolution enhancement in SDs with different Soluplus concentrations was found to be 20% < 40% < 60% < 70% < 90% < 80%, which was not totally the same as the order of solubility enhancement. This could be attributed to the fact that the hydrogen bonds affected more than the solubilization effects of Soluplus in the dissolution enhancement of some formulations.
Three reasons contributed to the high dissolution rate of SD. The enhanced aqueous solubility was regarded as the main factor for the dissolution improvement (reasons are mentioned in the section “Solubility study”). In addition, SIM in SD was in an amorphous or even molecular state, and there was no lattice energy for the amorphism, then the SDs did not need to overcome it throughout the dissolution process (Shamma & Basha, Citation2013). Moreover, the wettability of the drug was improved by Soluplus, which functioned a lot in enhancing the dissolution.
In vivo performance
Bioavailability studies were performed in beagle dogs to investigate whether the SIM/Soluplus SD increased the bioavailability. After oral administration, SIM turned into the SIM metabolite simvastatin acid (SIMA), a potent inhibitor of HMG-CoA reductase. Therefore, the plasma profiles of SIM and SIMA were compared after oral administration of the commercial tablet and SIM-SD-T (Luo, 2013).
The mean plasma concentrations versus time curves of SIM and SIMA following a single dose of each formulation are shown in ), respectively. There was an obvious difference between SIM-SD-T and the commercial tablet, indicating the difference in the rate and extent of SIM adsorption. The bioavailability parameters in and showed that the AUC(0–t) of SIM in SIM-SD-T and the commercial tablet was 299.92 ± 95.57 and 185.41 ± 100.15 ng/ml·h, respectively; that of SIMA were 170.71 ± 40.39, 108.79 ± 52.64 ng/ml·h (), respectively. Compared with commercial tablets, the average relative bioavailability of SIM and SIMA were increased 1.6- and 1.7-fold, respectively.
Figure 9. The mean plasma concentration-time curves of SIM (a) and SIM (b) in beagle dogs after oral administration (dose 20 mg, n = 6).
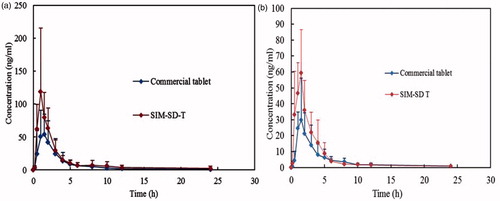
Table 4. SIM bioavailability parameters of SIM-SD-T and the commercial tablet in beagle dogs after oral administration (dose 20 mg, n = 6, means ± SD).
Table 5. SIMA* bioavailability parameters of SIM-SD-T* and the commercial tablet in beagle dogs after oral administration (dose 20 mg, n = 6, means ± SD).
The increased bioavailability could be explained by a combination of the following effects: (I) the Soluplus/SIM solid dispersion significantly improved the aqueous solubility and the dissolution rate of SIM; (II) the small particle size of SIM provided drug absorption with a large interfacial surface area, thereby enhancing the drug absorption in the gastrointestinal tract; (III) Soluplus and the drug formed micelles in vivo, which had a solubilization effect and the colloidal effect prevented the amorphous drug from undergoing devitrification. Moreover, the lower critical micelle concentration (CMC) of 7.6 mg/L in water (23 °C, data on file, BASF, Pharma Ingredients & Services, Germany; Technical Information 2009) made it easy to form stable colloidal micelles(Alam, Citation2012).
Preliminary stability test
Physical stability
Physical stability is always a challenging issue regarding amorphous drugs. In the study, the SIM/Soluplus SD of 80% Soluplus showed that little change took place in the dissolution profiles () and DSC thermogram () after 3 months (40 °C, RH 75%). The results showed that the SD was preliminary proved to be physical stable. However, the evidences were not full enough to illustrate the physical stability of the drug, as the low sensitivity of the methods. Actually, about 5–10% crystallinity cannot be detected by DSC.
Figure 10. Dissolution profile of solid dispersions (SD) containing 80% Soluplus after storage (40 °C, RH 75%).
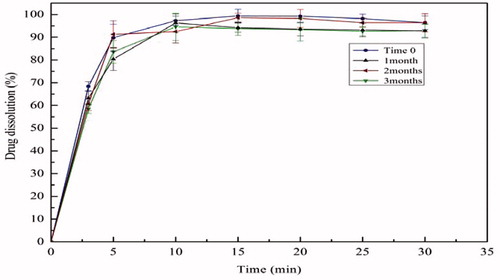
Figure 11. DSC thermograms of solid dispersions (SD) containing 80% Soluplus after storage (40 °C, RH 75%): (a) initially; (b) 1 month; (c) 2 months; (d) 3 months.
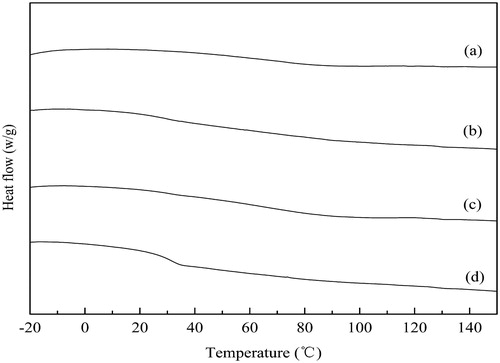
The physical stability was expected to be enhanced by the combined effect of the enhanced Tg value by Soluplus (Yonemochi & Inoue, Citation1999; Priemel, Citation2013) and the intermolecular hydrogen bonding (stated in Interactions between the amorphous SIM and Soluplus). But the existance of water was inevitable, which would lower the Tg.
Chemical stability
To date, very limited studies have been conducted on the chemical stability of the solid dispersion. In the study, the freshly prepared SIM/Soluplus SD sample showed a small amount of decomposition at about 1.8 ± 0.3%, and after 3 months (40 °C, RH 75%), higher amount of decomposition was detected at about 2.6 ± 0.5%. The corresponding PM had a rise in the amount of decomposition from 1.2 ± 0.2% to 2.4 ± 0.8%.
From the structure of SIM (), SIM was found to easily decompose in some environments, such as in humid, alkaline or aerobic environments (Ambike et al., Citation2005). It is believed that higher molecular mobility could accelerate chemical reactivity during storage (Djuris, Citation2013). There were many studies about the instability of amorphous simvastatin. Zhang (Citation2009) have reported that the Quench-cooled amorphous SIM after 42 days of storage has a significant decomposition amount at about 8.5 ± 1.3% and 9.6 ± 0.7%.
Therefore, changes occurred in the SIM/Soluplus SD was not dramatic during the storage period, compared with the PM. Extruded Soluplus, as a carrier, played an important role in stabilizing the SD. The results in the section “Powder X-ray diffraction” showed that SIM dispersed homogeneously in Soluplus, so the drug molecules were entrapped in the frozen polymers matrix. And also, Soluplus is not very hygroscopic (Zhang, Citation2013). Therefore, the surrounding thick Soluplus can protect SIM from moisture and oxide in the air, to some extent. Moreover, as the pH value of Soluplus was 4.86 (2.5 g Soluplus dissolved in 10 ml water), Soluplus was presumed to provide SIM with an acid environment, which helps preserve its chemical stability.
Conclusion
From the results of this study, amorphous SIM/Soluplus SD was found to enhance both the in-vitro and in-vivo performance. What’s more important, the SD displayed relatively good physicochemical stability during a 3-month storage period.
In conclusion, not only can Soluplus/SIM SD increase the bioavailability, but also displayed good physicochemical stability. Thus, the solid dispersion with Soluplus would be a more effective and stable oral delivery system for poorly water-soluble SIM.
Acknowledgements
Dr David B. Jack is gratefully thanked for correcting this article. The authors are thankful to BASF (Shanghai, China) for providing the gift sample of Soluplus to support this study.
Declaration of interest
The authors report no declarations of interest.
References
- Ahlneck C, Waltersson JO. (1986). Factorial designs in pharmaceutical preformulation studies. II. Studies on drug stability and compatibility in the solid state. Acta Pharm Suec 23:139–50
- Ahuja N, Katare OP, Singh B. (2007). Studies on dissolution enhancement and mathematical modeling of drug release of a poorly water-soluble drug using water-soluble carriers. Eur J Pharm Biopharm 65:26–38
- Alam MA. (2012). Solid dispersions: a strategy for poorly aqueous soluble drugs and technology updates. Expert Opin Drug Deliv 9:1419–40
- Ambike AA, Mahadik KR, Paradkar A. (2005). Spray-dried amorphous solid dispersions of simvastatin, a low tg drug: in vitro and in vivo evaluations. Pharm Res 22:990–8
- Avachata A, Rauta V, Avachat A. (2012). Solubility and dissolution enhancement of Nebivolol hydrochloride 2 using hydrophilic carriers 3. Asian J Pharma Sci 7:337–45
- Barmpalexis P. (2013). Development of PVP/PEG mixtures as appropriate carriers for the preparation of drug solid dispersions by melt mixing technique and optimization of dissolution using artificial neural networks. Eur J Pharm Biopharm 85:1219–31
- Celestino MT. (2012). Rational use of antioxidants in solid oral pharmaceutical preparations. Brazil J Pharma Sci 48:405–15
- Chokshi RJ. (2007). Improving the dissolution rate of poorly water soluble drug by solid dispersion and solid solution-pros and cons. Drug Deliv 14:33–45
- Dexia L. (2013). PLA/PEG-PPG-PEG/Dexamethasone implant prepared by hot-melt extrusion for controlled release of immunosuppressive drug to implantable medical devices, part 2: in vivo evaluation. Drug Deliv 20:134–42
- Djuris J. (2013). Effect of composition in the development of carbamazepine hot-melt extruded solid dispersions by application of mixture experimental design. J Pharm Pharmacol 66:232–43
- Greenhalgh DJ, Williams AC, Timmins P, York P. (1999). Solubility parameters as predictors of miscibility in solid dispersions. J Pharm Sci 88:1182–90
- Hong S, Shen S, Tan DCT, et al. (2014). High drug load, stable, manufacturable and bioavailable fenofibrate formulations in mesoporous silica: a comparison of spray drying versus solvent impregnation methods. Drug Deliv 22:1–12
- Hu R, Zhu J, Chen G. (2006). Preparation of sustained-release simvastatin microspheres by the spherical crystallization technique. Asian J Pharm Sci 1:47–52
- Jijun F. (2011). Nimodipine (NM) tablets with high dissolution containing NM solid dispersions prepared by hot-melt extrusion. Drug Dev Ind Pharm 37:934–44
- Jun SW. (2007). Preparation and characterization of simvastatin/hydroxypropyl-beta-cyclodextrin inclusion complex using supercritical antisolvent (SAS) process. Eur J Pharm Biopharm 66:413–21
- Kadam Y. (2011). Micelles from PEO-PPO-PEO block copolymers as nanocontainers for solubilization of a poorly water soluble drug hydrochlorothiazide. Colloids Surf B Biointerfaces 83:49–57
- Kalivoda A, Fischbach M, Kleinebudde P. (2012). Application of mixtures of polymeric carriers for dissolution enhancement of fenofibrate using hot-melt extrusion. Int J Pharm 429:58–68
- Linn M, Collnot EM, Djuric D. (2012). Soluplus as an effective absorption enhancer of poorly soluble drugs in vitro and in vivo. Eur J Pharm Sci 45:336–43
- Lobmann K. (2012). Co-amorphous simvastatin and glipizide combinations show improved physical stability without evidence of intermolecular interactions. Eur J Pharm Biopharm 81:159–69
- Luo Y, Xu L, Tao X, et al. (2013). Preparation, characterization, stability and in vitro-in vivo evaluation of pellet-layered Simvastatin nanosuspensions. Drug Dev Ind Pharm 39:936–46
- Mididoddi PK, Repka MA. (2007). Characterization of hot-melt extruded drug delivery systems for onychomycosis. Eur J Pharm Biopharm 66:95–105
- Pandya P. (2008). Co-solvent evaporation method for enhancement of solubility and dissolution rate of poorly aqueous soluble drug simvastatin: in vitro-in vivo evaluation. AAPS PharmSciTech 9:1247–52
- Priemel PA. (2013). Inhibition of surface crystallisation of amorphous indomethacin particles in physical drug-polymer mixtures. Int J Pharm 456:301–16
- Sarode AL. (2013). Supersaturation, nucleation, and crystal growth during single- and biphasic dissolution of amorphous solid dispersions: polymer effects and implications for oral bioavailability enhancement of poorly water soluble drugs. Eur J Pharm Biopharm 86:351–60
- Shamma RN, Basha M. (2013). Solupluse (R): a novel polymeric solubilizer for optimization of Carvedilol solid dispersions: formulation design and effect of method of preparation. Powder Technol 237:406–14
- Silva TD. (2010). Preparation and characterization of solid dispersion of simvastatin. Drug Dev Ind Pharm 36:1348–55
- Sousa M. (2010). Dynamical characterization of a cellulose acetate polysaccharide. J Phys Chem B 114:10939–53
- Srinarong P, de Waard H, Frijlink HW. (2011). Improved dissolution behavior of lipophilic drugs by solid dispersions: the production process as starting point for formulation considerations. Expert Opin Drug Deliv 8:1121–40
- Sun Y, Rui Y, Wenliang Z. (2008). Nimodipine semi-solid capsules containing solid dispersion for improving dissolution. Int J Pharm 359:144–9
- Surampalli G, Nanjwade BK. (2014). Novel tablet formulation of amorphous candesartan cilexetil solid dispersions involving P-gp inhibition for optimal drug delivery: in vitro and in vivo evaluation. Drug Deliv 31:1–15
- Tajber L, Corrigan OI, Healy AM. (2005). Physicochemical evaluation of PVP-thiazide diuretic interactions in co-spray-dried composites – analysis of glass transition composition relationships. Eur J Pharm Sci 24:553–63
- Telang C, Mujumdar S, Mathew M. (2009). Improved physical stability of amorphous state through acid base interactions. J Pharm Sci 98:2149–59
- Yonemochi E, Inoue Y. (1999). Differences in crystallization behavior between quenched and ground amorphous ursodeoxycholic acid. Pharm Res 16:835–40
- Yu H. (2013). Supersaturated polymeric micelles for oral cyclosporine a delivery. Eur J Pharm Biopharm 85:1325–36
- Zhang F. (2009). Influence of particle size and preparation methods on the physical and chemical stability of amorphous simvastatin. Eur J Pharm Biopharm 71:64–70
- Zhang K. (2013). Increased dissolution and oral absorption of itraconazole/Soluplus extrudate compared with itraconazole nanosuspension. Eur J Pharm Biopharm 85:1285–92