
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background information: Methotrexate (MTX), an anticancer drug of choice, has poor permeability across blood-brain barrier (BBB) making it unsuitable for brain tumor application. Its brain availability and scope of application was improved by preparation of reversible conjugate with lysine by capitalizing the endogenous transport system of lysine at BBB.
Methods: To enhance its delivery to brain, MTX was reversibly conjugated with l-Lysine by an amide linkage. It was characterized by advanced spectroscopy techniques including IR, NMR and MS. Furthermore, conjugate was assessed for stability, toxicity and drug release ability. In vivo distribution studies were done by radioscintigraphy study using 99mTc radioisotope.
Results: The structure of prodrug was confirmed by 1H-NMR, 13C-NMR and Mass. The m/e (mass to charge ratio) fragment was found at [M + H] 711.32 in Mass spectra. Stability and metabolic studies suggested that conjugate was stable at physiological pH (in Phosphate buffer pH 7.4 t1/2 is 70.25 ± 2.17 h and in plasma t1/2 is 193.57 ± 2.03 min) and circulated adequately to release MTX slowly in brain. In vivo biodistribution study showed that prodrug significantly increased the level of MTX in brain when compared with pharmacokinetic parameter of parent drug.
Conclusion: The brain permeability of MTX was enhanced significantly by this conjugate.
Introduction
The limited success of chemotherapy against brain tumors is primarily attributed to low Central Nervous System (CNS) penetration of antineoplastic agents across the blood-brain barrier (BBB). Methotrexate (MTX), a folate antagonist that interrupts the DNA synthesis has been widely used in many forms of cancer. Its brain penetration is poor at conventional doses (<100 mg/m2) and has to be administered in high-doses (1 g/m2 to 8 g/m2) to attain the desirable concentration in the brain (O’Brien et al., Citation2000; DeAngelis et al., Citation2002; Batchelor et al., Citation2003). However, being a folate analogue, it competes with natural folates leading to adverse effects and toxicity which get pronounced in higher doses limiting its application (West, Citation1997; Widemann & Adamson, Citation2006; Kozub & Simaljakova, Citation2011; Holmboe et al., Citation2012).
Considering the complexities of BBB and limitations of MTX, several approaches have been adapted for its brain delivery, including cetuximab (IMC-C225) dendrimer bio conjugates, osmotic BBB disruption, intracarotid administration of short-chain alkylglycerols and transnasal delivery (Neuwelt et al., Citation1981; Erdlenbruch et al., Citation2003; Wu et al., Citation2006; Shingaki et al., Citation2010). However, these methods have not been able to address many issues successfully and there is scope for evaluation of other approaches to enhance its brain delivery.
One approach that has recently received attention is reengineering of drugs based on the knowledge of the endogenous amino acid transportation system within the BBB for brain transport (Gabathuler, Citation2010; Pardridge, Citation2012; Peura et al., Citation2013). Amino acids crossing the BBB actively have the potential to be used for carrier-mediated transport of molecules across the brain. Many dietary amino acids display brain uptake utilizing a number of different transport systems that are broadly categorized as being either dependent or independent on Na+ (Oldendorf, Citation1971).
Although, MTX itself possess a glutamic acid moiety, its active brain uptake is poor. This is mainly because transport capacity of anionic amino acid system (system xG-) is quite low compared to neutral amino acids (Drewes et al., Citation1977; Al-Sarraf et al., Citation1995, Citation1997a,Citationb; Hawkins et al., Citation1995). Accordingly for enhanced brain uptake MTX can be conjugated to suitable amino acid carrier. With this concept, we have previously demonstrated better brain availability by glutamine conjugate of MTX (Subudhi & Singh, Citation2014). Unlike glutamic acid, lysine shares a common cerebrovascular cationic transporter (y+L system) with in situ Km values of 70 μM (O'Kane et al., Citation2006). It transports cationic amino acids in a Na+-independent manner and neutral amino acids in a Na+-dependent manner (White, Citation1985; Deves et al., Citation1992). Recently, lysine was used to enhance brain uptake of ketoprofen in rats. Although neither lysine nor ketoprofen was L-type amino acid transporter-1 (LAT-1)-substrates, their conjugate was found to have good affinity for LAT1 (Gynther et al., Citation2010). Keeping this in view, in the present study, we designed a reversible methotrexate-lysine conjugate (MTX-LYS) to enhance brain availability of MTX.
Materials and methods
Synthesis material and methods
All the reactions were performed with reagents of commercial high purity without further purification. Reactions were monitored by thin-layer chromatography. Purifications of the compounds were performed by crystallization and column chromatography. Purities of the final compound were determined to be >95% by an analytical HPLC (Young Lin Liquid Instrument, Hogye-dong, Anyang, Korea) on a promocil C18 column (4.6 mm × 250 mm, 5 µm). The FTIR spectrum was recorded on IR solution Version Affinity 1 (Shimadzu, Nishinokyo-Kuwabaracho, Nakagyo-ku, Kyoto, Japan). 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance II 400 spectrometer (Bruker Biospin, Fällanden, Switzerland) operating at 400.136 MHz and 100.61 MHz, respectively, using tetramethylsilane as an internal standard. The final products were also characterized by mass spectroscopy with Q-Tof micromass spectrometer (Waters, Milford, MA) equipped with an electrospray ionization source. Elemental analysis was carried out on a 2400 series II CHNS/O analyzer, Perkin Elmer, Winter Street Waltham, MA 02451 US, value found were within ±0.5% of theoretical ones. 99mTc-pertechnetate was provided by INMAS, Delhi, India. The radioactivity counter was a gamma ray counter (Type GRS23C, serial no. 458-425 Electronics Corporation of India Limited, Hyderabad, India). Imaging of animal was performed using Single Photon Emission Computerized Tomography (SPECT, LC 75-005, Diacam, Siemens AG, Erlanger, Germany) gamma camera. Animal handling and experimentation was carried out as per the guideline of the Institutional Animal Ethical Committee.
Synthesis procedure and characterization data
2,6-Bis((tert-butoxycarbonyl)amino)hexanoic acid (1) was prepared using di-tert-butyl dicarbonate following reported method with little modification (Keller et al., Citation1990; Martin et al., Citation2002; Laulloo et al., Citation2007). Compound 1 (1 g, 3 mmol) was dissolved in dimethyl formamide (10 mL). To this solution dicyclohexylcarbodiimde (0.7 g, 3 mmol, 1.1 eq.) dissolved in dimethyl formamide were added for 5 min with stirring. The stirring was continued at 0 °C for 2 h and then the solution of methotrexate (MTX) (1.36 g, 3 mmol, 1 eq.) in anhydrous dimethyl formamide (10 mL) was added drop wise. The resulting reaction mixture was stirred for 24 h at room temperature. Ethyl acetate (50 mL) was added to the reaction mixture and then filtered to remove the precipitated N,N-dicyclohexylurea (DCU). The clear filtrate was extracted with (3 × 20 mL) of 0.1 N HCl, (2 × 20 mL) of water, (2 × 20 mL) of brine, (3 × 20 mL) of saturated aqueous solution of sodium bicarbonate and (2 × 20 mL) of brine. The solution was then dried over anhydrous sodium sulphate and concentrated in vacuum. The residue was purified by column chromatography on silica gel using ethyl acetate/hexanes (70:30) to provide 1.34 g of compound 2.
Compound 2 (1.1 g, 1 mmol) was dissolved in dimethyl formamide, and trifluro acetic acid (TFA) 6 mL was added at 0 °C for 5 min with continuous stirring and kept at room temperature for 1.5 h. Then drying under vacuum to remove the TFA completely, recrystallization was carried out using ethyl acetate/diethyl ether mixture (1:10). The precipitate was filtered off, washed with cold diethyl ether and solvent was removed under vacuum to obtain the desired product as a light yellow solid with 0.38 g yield of compound 3 (MTX-LYS).
Light yellow solid, yield: 55%, m.p: 173.89 °C. Elemental analysis for C32H46N12O7 calculated C (54.07%), H (6.52%), N (23.65%) and found C (54.05%), H (6.56%), N (23.70%). FTIR (KBr), ν (cm−1): 3446.01 (O–H broad), 3310.63 (N–H asymm, str.), 3276.27 (N–H symm. str), 2927.34 and 2867.28 (C–H str), 1732.08 (C=O (OH), 1645.28 (C=O (NH) str), 1529.05 (C=N), 1475.51 (CH2, def), 1305.26 (C–H def). 1H–NMR (DMSO) δ (ppm): 11.08 (s, 1H, OH), 10.73 (s, 1H, OH), 8.58 (s, 4H, NH2), 8.36 (m, 4H, NH2), 8.23 (s, 1H, NH), 8.21 (s, 1H, NH), 8.17 (s, 1H, NH), 8.13 (s, 1H, CH (Pyrazine), 6.3–7.9 (m, Ar–H), 4.78 (s, 2H, CH2), 4.41–4.28 (t, 1H, CH), 3.73–3.75 (m, 1H, CH), 3.13–3.03 (m, 2H, CH2), 2.54–2.42 (t, 2H, CH2), 2.22–2.06 (m, 2H, CH2), 1.94–1.83 (m, 2H, CH2), 1.72–1.67 (m, 2H, CH2), 1.37–1.53 (m, 2H, CH2). 13C-NMR DMSO δ (ppm): 174.04 (–C(=O)OC), 173.81 (–C(=O)OC), 177.26 (–C(=O)NC), 177.39 (–C(=O)NC), 166.34 (–C(=O)NC), 162.64 (C(=N), NCPyrimidine), 162.34 (C(=N)CN, Pyrimidine), 154.47 (C(=N)C, Pyrazine), 150.82 C(=N)NN, Pyrimidine), 148.86 (C(=C)NC, Pyrazine), 146.14 (C(=C)NC, Benzene), 128.83 (C(=C)C, Benzene), 121.26 (C(=C)C, Benzene), 121.28 (C(=C)C, Benzene), 110.91 (C(=C)C, Benzene), 57.42 (C(N)CC), 57.39 (C(N)CC), 54.99 (C(N)CC), 51.81 (C(N)C), 42.02 (C(N)CH2), 40.18 (C(C)CH2), 39.97 (C(N)H3), 39.76 (C(N)CH2), 39.55 (C(N)CH2), 32.64 (C(C)CH2), 30.46 (C(C)CH2), 29.08 (C(C)CH2), 26.11 (C(C)CH2), 24.1 (C(C)CCH2), 24.08 (C(C) CCH2). MS (ESI+) m/z (%) (M+1): Calcd 711.36, Found 711.32.
In vitro studies
Stability studies in different buffer solutions
The MTX-LYS was investigated for their chemical stability in phosphate buffer solution at four pH values: 2.0, 4.9, 7.4 and 8.0 at 37 °C. Precisely, 1 mL methanol solution (0.140 µM/mL) of the conjugate was added into 4 mL of different buffer at 37 °C. After mixing, it was kept in a 37 ± 1 °C constant water bath and then 200 µL sample was withdrawn at different time points (0, 1, 2, 4, 8, 12 and 24 h). The disappearance of MTX-LYS was monitored by the HPLC method. The rate constant (Kdisapp, h−1) and half lives (t1/2, h) of the compounds in aqueous solution were calculated by linear regression of peak area against time in hour.
Stability in plasma extracts and brain homogenate
Blood was drawn from mice though orbital sinus and was collected in a heparinized tube paved with heparin sodium. Samples were centrifuged at 6000 rpm for 15 min to separate plasma, which was diluted with double volumes of water. The brain was removed and homogenized in cold phosphate buffer of pH 7.4 with proportion of 1:5 (w/v). Samples were then placed on ice and used immediately. One milliliter solution of MTX-LYS (0.140 µM/mL) in PBS 7.4 was added to plasma and brain homogenate, respectively, and gently vortexed. Samples were incubated at 37 °C and 200 µL aliquots were removed after 15, 30, 60, 90 and 120 min, respectively. Following centrifugation, the supernatants were analyzed by HPLC.
In vitro release of the MTX by MTX-LYS in brain homogenate
The MTX-LYS was treated in brain homogenate according to the procedure in the above section, and the peak of MTX was analyzed by HPLC. The pH was adjusted to acidic with 5 M HCl. Samples were incubated at 37 °C and 200 µL aliquots were removed after 15, 30, 60, 120, 180, 240 and 480 min, respectively. Samples were centrifuged for 15 min to remove proteins and the supernatants were analyzed by HPLC for determination of MTX. The mobile phase was composed of phosphate buffer (10 mmol dihydrogen phosphate and 10 mmol sodium hydroxide, pH 7.4) premixed, filtered and degassed buffer in water: acetonitrile (90:10) at a flow rate of 1 mL/min and the UV detector was set to monitor the signal at 310 nm corresponding to the maximum absorbance for MTX. Concentration was determined by normalizing peak integral area by dividing area for the peak of MTX-LYS at 15 min.
Distribution coefficients of the conjugates in 1-octanol/water
The distribution coefficient (log D) was determined by the shake flask method. The compound was dissolved in PBS 7.4 to a concentration of 1.40 µM/mL. The solution was carefully diluted with PBS 7.4, to obtain a series of known concentrations in order to create a calibration curve. The instrument was set to the maximum absorbance wavelength. The aqueous (PBS) solutions of MTX-LYS (1.40 µM/mL) stock solutions were prepared with corresponding volumes of 1-octanol (the ratio varied with the compounds). The mixture was kept for 24 h on a mechanical shaker and then allowed to stand long enough to separate the phases and achieve a saturation state. Concentration in the aqueous phase was determined by using UV-visible spectrophotometer. Based on a simple mass balance, concentration in the octanol phase was determined and the log D was calculated as the ratio of the concentration in the octanol phase to the concentration in the aqueous phase.
Protein binding study
Protein binding study was done following reported procedure (Rajput et al., Citation2012). Blood from goat was collected, heparinized and kept in deep freezer at 20 °C. Plasma was separated with aid of cooling centrifuge at 6000 rpm. Various concentrations of MTX and MTX-LYS (10, 20, 50 and 100 μg/mL) were prepared in plasma. Each dialyzing bag filled with 5 mL of plasma, containing a known amount of the drug, was then immersed in flask containing 5 mL of phosphate buffer 7.4 and the flasks were incubated at 37 °C for 24 h with orbital shaker at 50 rpm. At the end of the incubation period, the buffer as well as the contents of the dialyzing bags were analyzed separately by UV spectrophotometer and plasma protein binding was calculated.
Toxicological study
In vitro hemolytic toxicity study
Toxicity studies were carried out as per the guidelines of Organization of Economic Co-operation and Development (OECD-423) for testing of chemicals (Regd. No.-1283/c/09/CPCSEA). The degree of hemolysis was determined as per the reported method (Singhai et al., Citation1997). Human venous blood was collected using syringe prefilled with acid citrate dextrose buffer. The RBCs were separated from the whole blood by centrifugation (REMI, Mumbai, India) at 3000 rpm for 5 min. The supernatants and buffy coats were removed and discarded. The packed cell volume (PCV) was washed with normal saline and centrifuged again at 2000 rpm for 2 min. The 5 mL of PCV was then diluted to 100 mL with normal saline to get 5% RBC suspension. 0.5 mL of suitably diluted (0.1, 0.2, 0.3, 0.4%) plain MTX and MTX-LYS were added to 4.5 mL of normal saline and incubated for 1 h with RBC suspension. After centrifugation, supernatants were taken and diluted with an equal volume of normal saline and absorbance was measured at 540 nm. RBC suspension was added to 5 mL of saline and 5 mL distilled water, respectively, to obtain 0% and 100% hemolysis.
In vivo hematological studies
Healthy male albino rats of Sprague-Dawley strain of uniform body weight (100 ± 10 g) were selected for the study of hematological parameters. Fifteen animals were selected and divided into three groups comprised of three rats in each group. 25 and 250 mg/kg body weight of MTX and MTX-LYS were separately administered, intravenously into second and third groups of animals, respectively, daily up to 7 days. The first group was kept as control, which was maintained on same regular diet for 7 days. After 15 days, blood samples were collected from the animals of all the groups and analyzed for RBC count, WBC count, hematocrit (HCT), hemoglobin content, differential monocyte count, lymphocytes and neutrophils by pathology laboratory.
In vivo studies
Release of MTX from MTX-LYS in vivo
Mice were randomly divided into two groups, 24 in each group for different sampling time and housed in one cage. Each animal was injected with MTX or MTX-LYS in normal saline through the tail vein at a single dose equivalent to 7.15 µM/kg body weight of MTX. At appropriate time interval (15, 30, 60, 90, 120, 150, 180 and 240 min), the animal was sacrificed and 1 mL blood samples withdrawn from cardiac puncture were collected in heparinized tube. Plasma was immediately separated by centrifugation and diluted with PBS 7.4 to 1:3(v/v) which was stored at −20 °C until assay. Meanwhile, the brain sample was removed, weighed. Each tissue sample was homogenized and diluted with PBS 7.4 to 1:3 (w/v). The homogenates were also stored at −20 °C until assay. Before analyzing conjugate, hydrolysis was performed. After deproteinization, the mixture was centrifuged at 15 000 rpm for 15 min. A total of 200 µL aliquots were withdrawn, 200 µL of acetonitrile was added to each aliquot and vortexed. Samples were centrifuged for further 15 min to remove residual proteins and the supernatants were analyzed by the HPLC method. Concentration was determined as described in the section “In vitro release of the MTX by MTX-LYS in brain homogenate”.
Radiolabeling of MTX and MTX-LYS with 99mTc and optimization
The radiolabeling of MTX and MTX-LYS conjugates was done by dissolving the 2.0 mg equivalent of drug in 1 mL water for injection in a sterile glass vial. Then followed by an addition of 150–100 µg of a reducing agent, stannous chloride (1 mg/mL solution made in 1 N HCl) and pH was adjusted at 7.0 using 0.5 M sodium bicarbonate solution. To the resulting mixture (filtered through 0.22 µm membrane filter), 1–2 mL of 74 MBq 99mTc-pertechnetate containing 2.0 to 3.0 mCi was added drop wise and the reaction mixture was incubated at 25 ± 5 °C for 15–20 min and checked for radiolabeling efficiency by the thin layer chromatography method (Reddy et al., Citation2004). The procedure involved spotting 2 µL samples of radiopharmaceuticals on to chromatographic strip 10 cm in length. After developing in the solvent, the strip was cut into two portions (top:bottom: 1:3) and activity in each portion was measured in the form of count using gamma scintillation counter. 99mTc-labeled MTX and 99mTc-labeled MTX-LYS remained at the origin and free technetium travelled with the solvent front. The radiolabeling yield was expressed as a percentage of the total amount of radioactivity applied in the testing system. Percentage of colloid was determined using pyridine:acetic acid:water (3:2.5:1) as the mobile phase. The radiolabeling efficiency was calculated using established equation (Babbar et al., Citation2000). Radiochemical impurity that is likely to exist in the form of unconjugated technetium in 99mTc-labeled MTX-LYS conjugates and 99mTc-labeled MTX solution was determined by the instant thin layer chromatography-silica gel (ITLC–SG) strips as stationary phase. The effects of incubation time, pH and stannous chloride concentration on labeling were studied to achieve optimum reaction conditions. The in vitro stability of radiolabeled formulation was evaluated in 0.9% (w/v) sodium chloride and in mice plasma. After the optimization and evaluation, stable radiolabeled-drug conjugates of MTX were used for biodistribution study in mice.
Biodistribution study and radioscintigraphy imaging in mice
Swiss albino mice (2–3 month old of either sex) weighing 22–30 g of BALB/c strain were used for biodistribution studies. Animal were procured from the National Center of Disease Control, Sham Nath Marg, Delhi, India. The animals were selected at random from the stock colony maintained in the animal house facility. The animals were reared on laboratory chow pallets, fed ad libitum and had free access to food and water at all the time. The room was maintained at 25 ± 2 °C with natural daytime light and no light after 12 h until morning.
Three mice for each drug and conjugate per time point (15 min, 30 min, 60 min and 120 min) were used in the study. 3.7 MBq of radiolabeled compound containing 2 mg of MTX and MTX-LYS (equivalent to 2.81 µM/kg body weight) was injected through the tail vein of each mouse. The mice were killed humanely at different time intervals and the blood was collected using cardiac puncture. Subsequently, brain, lungs, heart, liver, kidney, spleen, intestine and stomach were dissected, washed twice using normal saline, made free from adhering tissue/fluid, and weighed. Radioactivity present in each tissue/organ was counted using shielded well-type gamma scintillation counter and expressed as % of radioactivity in blood to tissue relative distribution.
Gamma scintigraphy imaging was performed on mice following i.v. administrations of radiolabeled drug and its conjugate to determine the localization of drug in brain. 18.5 MBq of radiolabeled compound containing 2 mg of MTX and MTX-LYS (equivalent to 2.81 µM/kg body weight) was injected through the tail vein of each mouse. The mice were anaesthetized using 0.4 mL ketamine (50 mg/mL) intramuscular injection and placed on the imaging board. Imaging was performed using a gamma scintillation camera.
Statistical analysis
All experiments were conducted at least in triplicate and results were expressed as mean ± SD. The AUC0–t, AUMC0–t and MRT were calculated by the non-linear Trapezoidal rule. Statistical (Graphpad Prism 5.0 demo version) evaluation was performed by unpaired t-test with 95% confidence interval. p < 0.01 was considered significant. The RE and CE were calculated to evaluate the brain targeting property of conjugate. The value of RE and CE were defined as following:
where, sample represented (conjugate) and control (MTX).
Results and Discussion
Chemistry
In the present investigation, we prepared 2-(4-(((2,4-bis(2,6-diaminohexan amido)pteridin-6-l)methyl)(methyl)amino)benzamido)pentanedioic acid (MTX-LYS) as shown in Scheme 1. t-Butyloxycarbonyl (t-Boc) group was used as protecting group for the amine group of lysine. The pKa values of ɛ-amino and α-amino group are in the range of 10.5 to 9.5. So the reaction condition was maintained above pH 8.5 using NaOH solution (Smith, Citation2006). The use of carbodiimides, such as dicyclohexylcarbodiimide (DCC), for the activation of a carboxylic acid together with the addition of an amine is a frequently used procedure for amide bond formation in spite of troublesome formation and removal of urea by-products. The diimide moiety of DCC, which contains an electron-deficient central carbon atom, is attacked by the carboxylate and the highly reactive O-acylisourea is formed. The addition of an amine results in the formation of amide. We followed this procedure for synthesis of MTX-LYS. Considering the close nucleophilicity of both amino groups present on the pteridine nucleus of MTX, two amide bonds were formed with LYS. Following the bond formation amine group of lysine residue was deprotected by trifluoroacetic acid. The title compound was characterized by their respective IR, 1H-NMR, 13C-NMR and MS (Supplementary file). This is evident from the mass spectrum of MTX-LYS with molecular ion m/z peak at 711.32 (M + 1). This was further confirmed by fragment ion found at m/z 610.26 and 583.27. In 13C-NMR appearance of two close peaks at δ 177.26 and 177.39 for carbons for amides with LYS, in addition to peaks at δ 166.42 for amide of MTX and δ 174.04 and 173.81 for carbons in acid group of MTX. Peaks around δ 8–7 in the 1H-NMR spectrums support the proposed structure. Further confirmation of MTX-LYS was done with FTIR with peaks assigned to NH2 asymmetric and symmetric stretching at 3310.63 and 3276.27 cm−1. Peaks at 1732.08, 1645.28 (C=O str.) and 1529.05 (C=N str.) cm−1are also supportive of the proposed structure.
Scheme 1. Synthesis of MTX-LYS. Reagents and conditions: (i) NaOH, 1,4-Dioxane-Water, (BOC)2O, stir at room temp., 20 h; (ii) Anhydrous DMF, DCC, Ethyl Acetate, stir at 0 °C for 2 h, MTX stir at room temp. for 24 h; (iii) TFA, Anhydrous DMF, stir at room temp., 1.5 h.
Analytical method
The chromatographic separations were achieved on a promocil C18 column (4.6 mm × 250 mm, 5 µm), thermo stated at 27 °C. The solvent system comprised of phosphate buffer (10 mmol dihydrogen phosphate and 10 mmol sodium hydroxide, pH 7.4) premixed, filtered and degassed buffer in water:acetonitrile (90:10) was used as mobile phase at a flow rate of 1 mL/min. The retention time of MTX-LYS was 5.3 min (289 nm) and that of MTX was 7.3 min (310 nm). The method was validated before use as per the ICH guidelines for validation of analytical procedures. Calibration curves displayed good linearity (r2 > 0.995) within the tested concentration ranges (Supplementary file). The recovery range and the relative standard deviation for each of the analytes were found to be 98.5–99.52% and 0.48–1.29%, respectively.
Stability of conjugate in buffers
The pseudo first-order rate constants (Kdisapp) and half life (t1/2) of the conjugate are shown in . The compound appeared to be stable in pH 7.4 phosphate buffer and moderately stable at other pH. The slow hydrolysis at pH 7.4 indicated its ability for sustained release in physiological environment. Keeping in view the relative instability at pH other than this, it will not be suitable for oral administration. This for practical purposes can be considered as stable for further evaluation.
Table 1. Stability studies in different buffer solutions.
Metabolic study of conjugate
Metabolic stability
For a conjugate expected to reach brain must survive plasma transportation. MTX-LYS with amide bonds is expected to be slowly hydrolyzed in plasma (Rao et al., Citation1987; Simoes et al., Citation2009). Accordingly, the conjugate exhibited Kdisapp value of 3.58 × 10−3 min. This was faster than the degradation rate in pH 7.4 phosphate buffer suggesting contribution of enzymes in plasma to the degradation of conjugate. However, in spite of that stability in plasma is relatively reasonable to give enough time (t1/2 = 193.57 min) for distribution to brain. In brain homogenate, it gets degraded with a higher rate (). The decrease in stability in brain compared to plasma may suggest its hydrolysis and metabolism in brain which may be due to the metabolic activity of brain-specific forms of CYPs and amidases in brain (Rao et al., Citation1987; Voirol et al, Citation2000; Miksys & Tyndale, Citation2002). Similar observations have been reported in a recent work suggesting relatively higher metabolism in brain than plasma (Zhou et al., Citation2013). The degradation of conjugate in brain can be considered slow enough (t1/2 = 119.76 min) to favor a sustained action.
Table 2. Metabolic stability in plasma extracts and brain homogenate.
In vitro release of MTX in brain homogenate
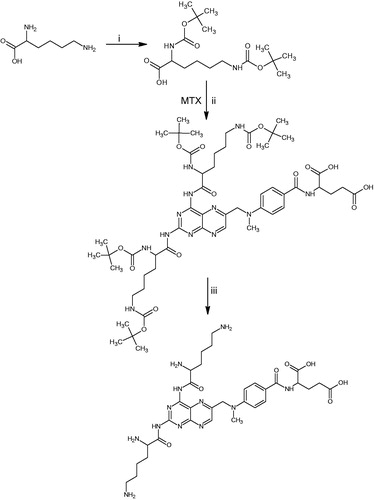
To study the release of MTX by the conjugate, peak corresponding to parent compound was analyzed. The data suggest that MTX was released from the conjugate steadily in brain homogenate (). The release of MTX is assumed to be by cleavage of amide bond. However, other than the two amide bonds produced by synthesis of conjugate, MTX itself contain one amide bond. The appearance of peak corresponding to standard MTX in HPLC shows that MTX is intact and only those two bonds of conjugate with LYS have undergone degradation. Although exact mechanism of this is not known, it is supported by the fact that hydrolysis of MTX at the amide bond is a very minor route of MTX metabolism. Only less than 5% of the drug gets eliminated as 2,4-diamino-N10-methylpteroic acid, because of hydrolysis by bacterial carboxypeptidases in intestinal tract (Donehower et al., Citation1979; Widemann et al., Citation2000).
Figure 1. Release of the MTX by MTX-LYS in brain homogenate. Values are expressed as mean ± SD (n = 3).
The release was observed to be slow (Kapp, 4.41 × 10−3). In contrast to disappearance of MTX-LYS (Kdisapp = 5.78 × 10−3), release of MTX was found to be less. This can be attributed to other degradation routes that do not result in MTX. However, the slow release can sustain the effects of MTX, which may be beneficial in the treatment of tumors that generally require a prolonged exposure (Ozeki et al., Citation2010). As the above in vitro studies demonstrated favorable features of the conjugate, we proceeded with the in vivo studies.
Lipophilicity and protein binding
Lipophilicity is one important parameter that influences the distribution of drug. A log D value of −1.34 for MTX-LYS indicated its polar nature. Conjugation to LYS, a relatively polar amino acid has not enhanced polarity, which is in accordance with a previous report on phenylalanine (Singh et al., Citation2009). However, the polarity of the conjugate (Supplementary file) can still be considered high enough to rule out possibility of passive diffusion across BBB.
Binding to plasma proteins strongly influences the entry of drugs into the central nervous compartments as in the presence of an intact barrier, only the plasma fraction unbound can freely penetrate (Norrby, Citation1985). The in vitro plasma protein binding study revealed that 38–43% of MTX was in bound form. This is in agreement with a previous study that showed plasma protein binding in the range of 25 to 55% (Combe et al., Citation1995). The conjugate exhibited 36–42% of plasma protein binding (Supplementary file). This suggests that both MTX and MTX-LYS exhibit comparable protein binding, and in general this level of binding can ensure enough free forms available for transport.
Toxicity study
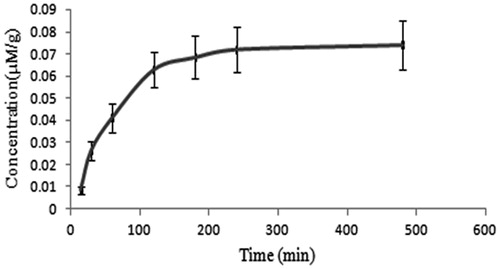
The MTX-LYS was well tolerated as suggested by the toxicity studies done. The hemolytic toxicity of MTX is a major limitation for its use. Thus, hemolytic toxicity was measured in terms of percent RBC hemolysis to monitor toxicity of the conjugate. MTX and conjugate exhibited concentration-dependent hemolysis, respectively. The conjugate exhibited relatively less hemolytic toxicity ().
Figure 2. Percent hemolysis with MTX and MTX-LYS. Values are expressed as mean ± SD (n = 3).
Further, to determine safety of the conjugate, hematological parameters were determined to study its effect on different components of blood. Blood samples were analyzed for RBC count, WBC count, hematocrit (HCT), hemoglobin (Hb) and differential lymphocytes count at a pathology laboratory. Although MTX-LYS in some cases exhibited less toxicity, the values were close enough to that of MTX (). This suggests that conjugate has not been able to mask the toxic profile of MTX, at the same time it has also not made it more toxic. Accordingly, for in vivo studies the conjugate dose profiles can be considered similar to that of MTX.
Table 3. Hematological parameters of animals treated with MTX and MTX-LYS conjugate at different doses (25 and 250 mg/kg) after 15 days.
In vivo studies
Pharmacokinetics in plasma and brain of MTX-LYS
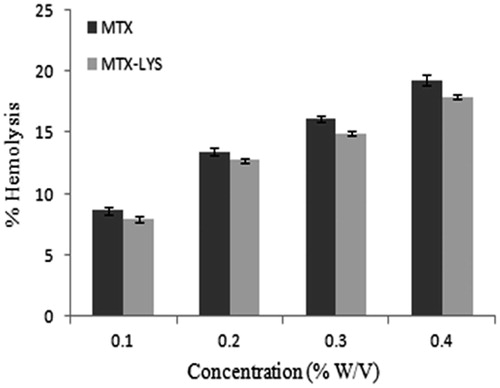
To understand the in vivo behavior of conjugate, we assessed the plasma pharmacokinetics of MTX and MTX-LYS. Pharmacokinetic parameters in blood were reported (). Free MTX of conjugate and MTX presented with an area under the concentration-time profile (AUC0–t) exhibited an increment of 57.18% compared to the parent drug. This indicated relatively prolonged plasma maintenance of the conjugate. The mean residence time (MRT) of MTX after administration of the conjugate increased by 73%. The half life of MTX from conjugate was 72.47% more than the parent drug. This suggested that it has helped increase its circulation time in plasma which may help the process of transportation across BBB. To further evaluate the possible transport of conjugate across BBB, distribution of MTX to brain from i.v. administration of conjugate and MTX was studied (). The AUC0–t and Cmax of MTX in brain after i.v. administration of conjugates were significantly higher than that after the injection of naked MTX. The relative uptake efficiency (RE) was 7.74 times for the conjugate. The concentration efficiency (CE) was 5.78 times for the conjugate. These results indicated that conjugate delivered MTX across BBB successfully ().
Figure 3. Concentration curve of MTX in brain after administration of MTX and MTX-LYS. Values are expressed as mean ± SD (n = 3).
Table 4. Pharmacokinetic parameter of MTX in plasma after administration of MTX and MTX-LYS.
Table 5. Pharmacokinetic parameter of MTX in brain after administration of MTX and MTX-LYS.
Biodistribution studies of MTX-LYS
The radiolabeling of MTX and MTX-LYS was done following an established method using 99mTc (Reddy et al., Citation2004; Vyas et al., Citation2006). The amount of SnCl2 2H2O, and incubation time for radiolabeling was optimized (). MTX-LYS labeled with 99mTc exhibited more than 97% radiolabeling efficiency. The radiochemical impurity that is likely to exist in the form of un-conjugated technetium was found to be 2.19% of MTX–LYS. The labeled conjugates were found to be stable in normal saline solution and in mice plasma up to 4 h (degradation <5% w/w) (). Thus, these formulations were found to be suitable for conducting radioscitigraphy studies in mice.
Table 6. Radiolabeling of MTX and MTX-LYS.
Table 7. In vitro stability of radiolabeled complexes of MTX and MTX-LYS in saline and plasma at different time intervals.
The stable radiolabeled MTX and MTX-LYS were used for bio-distribution study. It was injected (equivalent to 2.81 µM/kg body weight) through the tail vein of Swiss albino mice. Radioactivity present in each tissue/organ was measured using shielded well-type gamma scintillation counter as % radioactivity. Measurement and expression of exact bio-distribution to tissues is subjected to many limitations. Thus, tissue to blood ratio is an important parameter to indicate drug distribution (Poulin & Theil, Citation2000; Muller et al., Citation2004). Accordingly, blood label after 15 min of administration was considered 100% and relative distribution to other tissues expressed as percentage of it (). To determine transportation of conjugate from blood to brain, at each time point the level of MTX and MTX-LYS in brain was expressed as percentage of that of blood.
Table 8. Relative tissue distribution (%) of MTX and MTX-LYS.
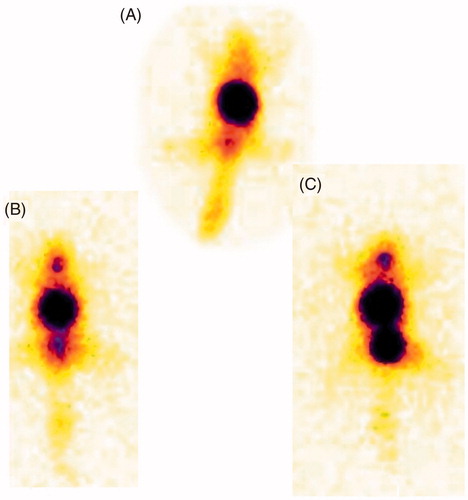
Considering the fact that MTX level in brain from both MTX and MTX-LYS reached highest level after 60 min of administration () and stability of radiolabeled compound (4 h), distribution data was compiled up to 120 min of administration. Both MTX and the conjugate were widely distributed to tissues including liver, spleen, lung and kidney as observed from radioscintigraph images () and radioactivity count (). The drug level reduced with time suggesting tissue up take and elimination. Distribution to heart tissue was less and comparable for both conjugate and the parent drug. The liver and spleen level of MTX and conjugate increased up to 30 min and then decreased with time. Distribution of both parent drug and conjugate was similar to stomach and intestine. Relatively low distribution of MTX-LYS to lung tissue suggests less fast-pass lung elimination of the conjugate. In contrast to this, higher distribution of conjugate to kidney suggests significance of its elimination by this route. However, distribution to brain tissue presents a very contrast figure. The relative brain distribution of MTX-LYS was much higher than that of MTX. This indicates effective transport of MTX-LYS across BBB. The polar nature of the conjugate cannot justify its higher brain penetration. The significant brain permeability suggests involvement of active transportation system similar to that of lysine-ketoprofen conjugate.
Figure 4. Gamma scintigraphy images of mice (A) MTX after 120 min, (B) MTX-LYS, after 60 min, (C) MTX-LYS after 120 min.
However, amino acid transporters are also expressed peripherally, but distribution of conjugate to these tissues are poor. This can be explained by the fact that amino acid transporters are selectively expressed at BBB (Boado et al., Citation1999) and peripheral LAT have less affinity for amino acids as compared to those at BBB (Pardridge et al., Citation1983; Miller et al., Citation1985; Pardridge & Hargreaves, Citation1988). Further, lysine and its conjugates are selectively uptaken by tumor cells (Perevoshchikova, Citation1960; Begleiter et al., Citation1979; Kaminskas et al, Citation2011). This conjugate can thus have better tumor penetration which may impart its tumor targeting property. Further work is going on to establish its transporter interaction and tumor selectivity studies.
Conclusion
In the present study, a novel lysine conjugate of MTX has been shown to increase the brain penetration of MTX. The chemical stability of MTX-LYS is sufficient for i.v. administration. Its protein binding is comparable to the parent drug, but it is more polar that limits its passive permeation. It is stable enough in the plasma to survive for brain transportation. The enzymatic release of MTX from MTX-LYS is slower both in vivo and in vitro, which can be an essential feature for sustained drug action. The peripheral tissue distribution of MTX-LYS is less but brain transport is more compared to MTX, which provides evidence of its suitability for selective brain transport. Further studies are ongoing in order to explore its transporter interaction and possible tumor uptake enhancement.
Supplementary material available online.
Supplemental Material.pdf
Download PDF (225.3 KB)Acknowledgements
The authors are thankful to Dr Anil Kumar Mishra, HOD and Joint Director, Division of Cyclotron and Radiopharmaceutical Sciences, Institute of Nuclear Medicine and Allied Sciences (INMAS), Delhi for his permission and Dr Krushna Chuttani, Scientist, INMAS for her help in carrying out radioscintigraphy experiments. We are also thankful to Sophisticated Analytical Instrumentation Facility, Panjab University, Chandigarh, for help in spectral characterization and IPCA laboratories, Daman, for providing methotrexate as gift sample.
Declaration of interest
There is no conflict of interest with any financial organization regarding the material discussed in the manuscript. The authors alone are responsible for the content and writing of the manuscript.
References
- Al-Sarraf H, Preston JE, Segal MB. (1995). The entry of acidic amino acids into brain and CSF during development, using in situ perfusion in the rat. Dev Brain Res 90:151–8
- Al-Sarraf H, Preston JE, Segal MB. (1997a). Acidic amino acid accumulation by rat choroid plexus during development. Dev Brain Res 102:47–52
- Al-Sarraf H, Preston JE, Segal MB. (1997b). Changes in the kinetics of the acidic amino acid brain and CSF uptake during development in the rat. Dev Brain Res 102:127–34
- Babbar AK, Singh K, Goel HC, et al. (2000). Evaluation of 99mTc labeled Photosan-3, a heamatoporphyrin derivative, as a potential radiopharmaceutical for tumor scintigraphy. Nucl Med Biol 27:419–26
- Batchelor T, Carson K, O’Neill A, et al. (2003). Treatment of primary CNS lymphoma with methotrexate and deferred radiotherapy: a report of NABTT 96–07. J Clin Oncol 21:1044–9
- Begleiter A, Lam H, Grover J, et al. (1979). Evidence for active transport of melphalanby amino acid carriers in L5178Y lymphoblasts in vitro. Cancer Res 39:353–9
- Boado RJ, Li JY, Nagaya M, et al. (1999). Selective expression of the large neutral amino acid transporter at the blood–brain barrier. PNAS 96:12079–84
- Combe B, Edno L, Lafforgue P, et al. (1995). Total and free methotrexate pharmacokinetics, with and without piroxicam, in rheumatoid arthritis patients. Br J Rheumatol 34:421–8
- DeAngelis LM, Seiferheld W, Schold SC, et al. (2002). Combination chemotherapy and radiotherapy for primary central nervous system lymphoma: radiation therapy. Oncology Group Study 93-10. J Clin Oncol 20:4643–8
- Deves R, Chavez P, Boyd CA. (1992). Identification of a new transport system (y + L) in human erythrocytes that recognizes lysine and leucine with high affinity. J Physiol 454:491–501
- Donehower RC, Hande KR, Drake JC, Chabner BA. (1979). Presence of 2,4-diamino-N10-methylpteroic acid after high-dose methotrexate. Clin Pharmacol Ther 26:63–72
- Drewes LR, Conway WP, Gilboe DD. (1977). Net amino acid transport between plasma and erythrocytes and perfused dog brain. Am J Physiol 233:320–5
- Erdlenbruch B, Schinkhof C, Kugler W, et al. (2003). Intracarotid administration of short-chain alkylglycerols for increased delivery of methotrexate to the rat brain. Br J Pharmacol 139:685–94
- Gabathuler R. (2010). Approaches to transport therapeutic drugs across the blood–brain barrier to treat brain diseases. Neurobiol Dis 37:48–57
- Gynther M, Jalkanen A, Lehtonen M, et al. (2010). Brain uptake of ketoprofen–lysine prodrug in rats. Int J Pharm 399:121–8
- Hawkins R, DeJoseph MR, Hawkins PA. (1995). Regional brain glutamate transport in rats at normal and raised concentrations of circulating glutamate. Cell Tissue Res 281:207–14
- Holmboe L, Andersen AM, Morkrid L, et al. (2012). High dose methotrexate chemotherapy: pharmacokinetics, folate and toxicity in osteosarcoma patients. Br J Clin Pharmacol 73:106–14
- Kaminskas LM, Kelly BD, McLeod VM, et al. (2011). Characterisation and tumour targeting of PEGylated polylysine dendrimers bearing doxorubicin via a pH labile linker. J Control Release 152:241–8
- Keller O, Keller WE, Look GV, Wersi G. (1990). Tert-butoxycarbonylation of amino acids and their derivatives: n-tert-butoxycarbonyl-l-phenylalanine organic syntheses coll. 7:70
- Kozub P, Simaljakova M. (2011). Systemic therapy of psoriasis: methotrexate. Bratisl Lek Listy 112:390–4
- Laulloo SJ, Khodabocus A, Hemraz UD, Sunasee R. (2007). Use of di-tert-butyl-dicarbonate both as a protecting and activating group in the synthesis of dipeptides. Synth Commun 37:4191–7
- Martin B, Posseme F, Barbier CL, et al. (2002). (Z)-1,4-Diamino-2-butene as a vector of boron, fluorine, or iodine for cancer therapy and imaging: synthesis and biological evaluation. Bioorg Med Chem 10:2863–71
- Miksys SL, Tyndale RF. (2002). Drug-metabolizing cytochrome P450s in the brain. J Psychiatry Neurosci 27:406–15
- Miller LP, Patridge WM, Braun LD. (1985). Kinetic constants for blood—brain barrier amino acid transport in conscious rats. J Neurochem 45:1427–32
- Muller M, delaPeria A, Derendrof H. (2004). Issues in pharmacokinetics and pharmacodynamics of antinfective agents: distribution to tissues. Antimicrob Agents Chemother 48:1441–53
- Neuwelt EA, Diehl JT, Vu LH, et al. (1981). Monitoring of methotrexate delivery in patients with malignant brain tumors after osmotic blood–brain barrier disruption. Ann Intern Med 94:449–54
- Norrby SR. (1985). Role of cephalosporins in the treatment of bacterial meningitis in adults. Overview with special emphasis on ceftazidime. Am J Med 79:56–61
- O’Brien P, Roos D, Pratt KL, et al. (2000). Phase II multicenter study of brief single-agent methotrexate followed by irradiation in primary CNS lymphoma. J Clin Oncol 18:519–26
- O’Kane RL, Vina JR, Simpson I, et al. (2006). Cationic amino acid transport across the blood-brain barrier is mediated exclusively by system y+. Am J Physiol Endocrinol Metab 291:412–19
- Oldendorf WH. (1971). Uptake of radiolabeled essential amino acids by brain following arterial injection. Proc Soc Exp Biol Med 136:385–6
- Ozeki T, Hashizawa K, Kaneko D, et al. (2010). Treatment of rat brain tumors using sustained-release of camptothecin from poly(lactic-co-glycolic acid) microspheres in a thermoreversible hydrogel. Chem Pharm Bull 58:1142–7
- Pardridge WM. (1983). Brain metabolism: a perspective from the blood–brain barrier. Physiol Rev 63:1481–535
- Pardridge WM. (2012). Drug transport across the blood–brain barrier. J Cereb Blood Flow Metab 32:1959–72
- Patridge WM, Hargreaves KM. (1988). Neutral amino acid transport at the human blood–brain barrier. J Biol Chem 263:19392–7
- Perevoshchikova KA. (1960). The correlation between the active concentration of labeled amino acids by the cells, and their inclusion in the proteins of tumors and normal tissues of experimental animals in vitro. Bull Exp Biol Med 49:162–5
- Peura L, Malmioja K, Huttunen K, et al. (2013). Erratum to: Design, synthesis and brain uptake of lat1-targeted amino acid prodrugs of dopamine. Pharm Res 30:1714–17
- Poulin P, Theil FP. (2000). A priori prediction of tissue: plasma partition coefficients of drugs to facilitate the use of physiologically-based pharmacokinetic models in drug discovery. Pharm Sci 89:16–35
- Rajput N, Dumka VK, Sandhu HS. (2012). Disposition kinetics and in vitro plasma protein binding of cefpirome in cattle. Vet Arhiv 82:1–9
- Rao TS, Baker GB, Coutts RT. (1987). N-(3-chloropropyl) phenylethylamine as a possible prodrug of beta-phenylethylamine: studies in the rat brain. Prog Neuropsychopharmacol Biol Psychiatry 11:301–8
- Reddy L, Sharma R, Chuttani K. (2004). Etoposide-incorporated tripalmitin nanoparticles with different surface charge: formulation, characterization, radiolabeling, and biodistribution studies. AAPS J 6:55–64
- Shingaki T, Inoue D, Furubayashi T, et al. (2010). Transnasal delivery of methotrexate to brain tumors in rats: a new strategy for brain tumor chemotherapy. Mol Pharmaceutics 7:1561–8
- Simoes MF, Valente E, Gomez MJ, et al. (2009). Lipophilic pyrazinoic acid amide and ester prodrugs stability, activation and activity against M. tuberculosis. Eur J Pharm Sci 37:257–63
- Singh AP, Ramadan WM, Dahiya R, et al. (2009). Product development studies of amino acid conjugate of aceclofenac. Curr Drug Deliv 6:208–16
- Singhai AK, Jain S, Jain NK. (1997). Evaluation of an aqueous injection of Ketoprofen. Pharmazie 52:149–51
- Smith GP. (2006). Kinetics of amine modification of proteins. Bioconjug Chem 17:501–6
- Subudhi BB, Singh VK. (2014). Development of reversible glutamine conjugate of methotrexate for enhanced brain delivery. Med Chem Res. DOI: 10.1007/s00044–014–1172–0
- Voirol P, Jonzier-Perey M, Porchet F, et al. (2000). Cytochrome P-450 activities in human and rat brain microsomes. Brain Res 855:235–43
- Vyas TK, Babbar AK, Sharma RK, et al. (2006). Preliminary brain-targeting studies on intranasal mucoadhesive microemulsions of sumatriptan. AAPS Pharm Sci Tech 7:49–57
- West SG. (1997). Methotrexate hepatotoxicity. Rheum Dis Clin North Am 23:883–915
- White MF. (1985). The transport of cationic amino acids across the plasma membrane of mammalian cells. Biochim Biophys Acta 822:355–74
- Widemann BC, Adamson PC. (2006). Understanding and managing methotrexate nephrotoxicity. Oncologist 11:694–703
- Widemann BC, Sung E, Anderson L, et al. (2000). Pharmacokinetics and metabolism of the methotrexate metabolite, 2,4-diamino-N10-methylpteroic acid. J Pharmacol Exp Ther 294:894–901
- Wu G, Barth RF, Yang W, et al. (2006). Targeted delivery of methotrexate to epidermal growth factor receptor–positive brain tumors by means of cetuximab (IMC-C225) dendrimer bioconjugates. Mol Cancer Ther 5:52–9
- Zhou K, Khokhar JY, Zhao B, Tyndale RF. (2013). First demonstration that brain CYP2D-mediated opiate metabolic activation alters analgesia in vivo. Biochem Pharmacol 85:1848–55