
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
As with many other hydrophobic anticancer agents, 20(S)-protopanaxadiol (PPD) has a very low oral bioavailability. In this study, a precipitation-combined ultrasonication technique was used to prepare PPD nanosuspensions. The mean particle size of the nanosuspensions was approximately 222 ± 12 nm, the drug payload achieved 50% after lyophilization and the maximum PPD concentration can reach 100 mg/ml, which is over 30 000 times the solubility of PPD in aqueous solution (3 μg/ml). After oral administration, the Cmax and AUClast values of PPD nanosuspensions were approximately 3.66-fold and 3.48-fold as those of PPD coarse suspensions, respectively. In contrast to the free drug solution, PPD nanosuspensions showed higher in vitro anti-tumor activity against HepG-2 cells (an IC50 value of 1.40 versus 5.83 μg/ml at 24 h, p < 0.01). The in vivo study in H22-tumor-bearing mice demonstrated that PPD nanosuspensions showed good anti-tumor efficacy with an inhibition rate of 79.47% at 100 mg/kg, while 50 mg/kg of cyclophosphamide was displayed as positive control, and the inhibition rate was 87.81%. Considering the highest drug payload, oral bioavailability reported so far, significant anti-tumor efficacy and excellent safety of encapsulated drugs, PPD nanosuspensions could be used in potential effective strategies for anticancer therapy; further investigation is ongoing.
Introduction
20 (S)-protopanaxadiol (PPD) is mainly derived from ginseng and is an aglycone of the PPD-containing ginsenosides, such as ginsenoside Rb1, Rb2, Rb3, Rh2, and Rg3. PPD saponins, such as Rg3 and Rh2, have been shown to possess a variety of beneficial effects for anti-tumor, antifatigue, antiaging, anticancer, and immune regulatory processes (Ota et al., Citation1991; Iishi et al., Citation1997; Li et al., Citation2009). Moreover, the pharmacological effects of PPD are similar to or stronger than those of Rh2 and Rg3 (Hasegawa et al., Citation2000; Lee et al., Citation2000; Popovich & Kitts, Citation2004). Previous studies indicated that PPD showed powerful pleiotropic anti-cancer effects in several cancer cell lines and the capability of inhibiting metastasis (Li et al., Citation2006; Liu et al., Citation2007; Dong et al., Citation2011; Zhu et al., Citation2011), thus becomes a potential therapeutic agent in the prevention and treatment of cancer (Popovich & Kitts, Citation2002; Wang et al., Citation2007; Yu et al., Citation2007; Usami et al., Citation2008). As such, it is currently undergoing phase I clinical trials in China. However, preclinical assessment of the pharmacokinetics of radiolabeled PPD in rats and dogs shows that the oral bioavailability of PPD is low because of poor oral absorption and extensive metabolism by cytochrome P450 (Li et al., Citation2011).
As such, some studies employed cubic nanoparticles (Jin et al., Citation2013a), lipid cubic nanoparticles (Jin et al., Citation2013b), drug–phospholipid complex nanoparticles (Xia et al., Citation2013) and liquid crystalline nanoparticles (Jin et al., Citation2013c) to resolve this problem, with the bioavailability increased by 69%, 66%, 116%, and 148%, in comparison to the raw PPD material; however, the drug payload of them is less than 20%.
Although we have also formulated PPD as emulsions, oil solution (Han et al., Citation2010), hydroxypropyl-β-cyclodextrin inclusion and pharmacosomes (Han et al., Citation2013) to resolve this problem, none of the resultant oral absolute bioavailability could reach 50% and none of the dug-loading content reached 25%. In order to obtain high drug-loaded formulation to further enhance oral bioavailability of PPD, the new carrier is required.
Nanosuspensions are colloidal drug delivery systems that contain drug particles and stabilizers. The mean size of the drug particles is in nanometer range, typically between 10 and 1000 nm (Keck & Müller, Citation2006; Gao et al., Citation2008), which could increase the solubility, dissolution rate, and mucosal adhesion.
In this article, PPD nanosuspensions were first studied, in which the drug in suspension is at a maximum concentration of 100 mg/ml, the drug-loading content achieved 50%, and the oral bioavailability of PPD was increased by 2.48 times (the absolute bioavailability was approximately 66%). In addition, the in vitro and in vivo anti-tumor activities on PPD were not reported so far, as thus the in vitro and in vivo anti-tumor activities were systematically evaluated in this study, whose results indicate that PPD nanosuspensions have great potential for the treatment of hepatic cancer.
Materials and methods
Materials
Shanghai Shengjia BioScientific Corp., Ltd. (Shanghai, China) provided the 20(S)-protopanaxadiol (PPD), which was characterized by greater than 99% purity. The bovine serum albumin (BSA) was purchased from Sigma Chemical (St. Louis, MO). The water used in the experiments was deionized, and all other organic solvents were of the highest commercially available grade.
Animals
Male Sprague–Dawley (SD) rats (200 ± 20 g) were obtained from the Experimental Animal Center of Peking University (Beijing, China). The female ICR mice (20 ± 2 g) were supplied by the Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). All the animals were acclimatized at a temperature of 25 ± 2 °C and a relative humidity of 70 ± 5% under natural light/dark conditions for 1 week and provided food and water ad libitum. All the experimental procedures were performed in accordance with the Guidelines for Ethical and Regulatory Use for Animal Experiments as defined by the Institute of Medicinal Plant Development (IMPLAD) in China.
Instrumentation and chromatographic conditions
The instrumentation system consisted of a HPLC system was an Agilent Model 1100 series (Agilent, Palo Alto, CA) consisting of a pump, autosampler, and column oven set at 22 °C. An API-4000 tandem mass spectrometer (Applied Biosystems/MDS SCIEX, Concord, Ontario, Canada) was equipped with an electrospray ionization (ESI) source. Analyst software (Applied Biosystems/MDS SCIEX, Foster City, CA, version 1.3) was installed on a DELL computer. Separation was carried out using MassLynx 4.1 software (MassLynx Software, Milford, MA) on an a C18 analytical column (Acclaim® RSLC 120 Å, 2.1 × 150 mm2, 2.2 µm, Dionex, Sunnyvale, CA). The isocratic HPLC mobile phase: 0.1 mM ammonium chloride–acetonitrile (10: 90, v/v); flow rate: 0.2 ml/min; the column temperature: 22 °C; injected volume: 20 µl. The mass spectrometer was operated in the positive ion mode. Detection by multiple reaction monitoring (MRM) used the transitions at m/z 461.2 → 425.4 for PPD; collision gas, curtain gas, gas 1 and gas 2 (nitrogen) 5, 10, 60 and 30 p.s.i., respectively; dwell time 200 ms; IonSpray voltage 5500 V; source temperature 200 °C; declustering potential (DP) 12 V; collision energy (CE) 12 eV. Low resolution was used for both Q1 and Q3 mass detection. The lower limits of detection and quantification were 6 and 20 ng, respectively. The calibration curve for PPD was established, and the correlation coefficient was greater than 0.99.
Preparation of nanosuspensions
PPD nanosuspensions were prepared by the precipitation–ultrasonication method. Bulk PPD powder was dissolved in acetone (25 °C) to form an organic solution containing 40 mg/ml of the drug. Then, 2.5 ml of the organic solution was slowly introduced into a 10 ml BSA solution (1%, w/v, PPD/BSA 1:1, w/w) at 25 °C under continuous sonication (250 W) (Ultrasonic cleaner, Kun Shan Ultrasonic Instruments Co., Ltd., Shanghai, China). The resultant suspensions were evaporated under vacuum at 40 °C until no residual acetone smell remained. Then, the PPD nanosuspensions were obtained, in which PPD concentration was 10 mg/ml. Each experiment was performed in triplicate.
Characterizations of nanosuspensions
Lyophilization
To further enhance the storage stability, PPD nanosuspensions were lyophilized before storage and subsequent use. PPD nanosuspensions were rapidly cooled to −80 °C for 12 h and then freeze-dried in an LGJ-10B freeze-dryer (Sihuan Laboratory Instruments Co., Ltd., Sichuan, China) under vacuum (pressure <10 pa) for 24 h. No cryoprotectant was needed during this procedure.
The sizes and zeta potentials determination
The mean particle size (z-average), polydispersity index (PDI), and zeta potential (ZP) were determined by dynamic light scattering (DLS) (Zetasizer nano ZS 90, Malvern Instruments, Malvern, UK). All the measurements were performed with a laser wavelength of 633 nm at 25 °C. Each sample (5 mg/ml) was measured in triplicate.
Morphology of nanosuspensions
The morphologies of bulk PPD power and PPD nanosuspensions were examined with scanning electron microscopy (SEM, JEM-1400, JEOL Ltd., Tokyo, Japan). SEM samples were glued and mounted on metal sample plates. The samples were gold coated with a sputter coater using an electrical potential of 2.0 kV at 30 mA for 240 s.
X-ray power diffraction measurements
X-ray powder diffraction (XRD) was performed on a Bruker D8 advance instrument controlled by Diffrac plus XRD commander software. Bulk PPD power, lyophilized PPD nanosuspensions, and physical mixture of PPD with BSA were scanned over an angular range of 3–40 °C of 2θ, with a step size of 0.02 ° and 2 s/step at 40 mA and 40 kV.
Differential scanning calorimetry (DSC) studies
DSC measurement was performed using a DSC instrument (DSC Q 200, TA Co., New Castle, DE). Samples of approximately 5 mg were placed in standard aluminum pans, sealed with a lid and run at a scanning rate of 10 °C/min over 30–310 °C under nitrogen.
In vitro cytotoxicity of nanosuspensions against HepG 2 cells
Cell culture
The human hepatoma cell line HepG 2 was a gift from the Chinese Academy of Medical Science (Beijing, China) and was cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% antibiotic solution (penicillin 100 U/ml and streptomycin 100 μg/ml) at 37 °C in a humidified 5% CO2 incubator.
MTT assay
The HepG 2 cells in the logarithmic growth-phase were collected and seeded in a 96-well plate (7 × 103 cells/well) for 24 h. The cells were treated with PPD nanosuspensions or PPD DMSO solution (a final concentration of 0.625, 1.25, 2.5, 5, and 10 µg/ml) for 12, 24, and 36 h, with RPMI 1640 as the negative control. Each sample or control group contained six parallel wells. An MTT assay was performed by adding 20 μl of the MTT working solution (5 mg/ml, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; Sigma, St. Louis, MO) to each well and then incubated for an additional 4 h, after which the culture media were replaced with 150 μl of DMSO. The light absorbance was measured at 570 nm. The cell inhibition rate was calculated according to the following formula:
The cell cytotoxicity was expressed as IC50 (the concentration of a drug that is required for 50% inhibition rate relative to the controls, which was estimated from a linear regression analysis of the experimental data).
Pharmacokinetics and bioavailability in rats
Formulation
Bulk PPD powder was dispersed in 0.9% (w/v) physiological saline to produce coarse suspensions (5 mg/ml), the resulting suspensions were subjected to continuous ultrasound (Kunshan Ultrasonic Instruments Co., Ltd., Kunshan, China) for 10 min for intragastric (i.g.) administration.
PPD solution of 5 mg/ml was prepared by dissolution of PPD in DMSO and then sequential additions of Tween 80 and physiological saline (DMSO:Tween 80:physiological saline 0.5:0.5:9, v/v/v) for intravenous (i.v.) administration.
The freeze-dried PPD nanosuspensions were reconstituted in 0.5 M PBS (pH 8.0) (5 mg/ml) before i.g. administration and in 0.9% NaCl solution (5 mg/ml) before i.v. administration.
Pharmacokinetics and bioavailability studies in rats
The SD rats were randomly divided into five groups (six rats per group). PPD nanosuspensions and coarse suspensions were i.g. administered, while PPD nanosuspensions and solution were i.v. administered at a dose of 25 mg/kg. The rats were fasted for 24 h before dosing. Water was freely accessible; no food was allowed until the rats had been given the drug for 6 h. Blood samples were collected by orbital sinus bleeding into heparinized test tubes at 0 (per-dose), 0.083, 0.167, 0.25, 0.5, 0.75, 1, 1.5, 2, 4, 6, 8, 12, and 24 h after the dosing. After centrifugation at 5000 rpm for 5 min, the plasma was separated and stored at −80 °C until the analysis.
Sample preparation method
An aliquot of 200 μl of plasma was accurately transferred to a clean 5.0 ml test tube, 10 μl of finasteride solution (10 μg/ml in acetonitrile) as the internal standard was added and mixed well, followed by the addition of 3 ml of dichloromethane. After vortex mixing for 10 min, the mixture was kept for 10 min at room temperature and then centrifuged at 10 000rpm for 5 min. The organic layer was collected and evaporated to dryness under nitrogen at 40 °C. The residues were dissolved in 200 µl of acetonitrile and centrifuged at 3000 rpm for 10 min, and 20 µl of supernatant was analyzed by HPLC (Agilent, Palo Alto, CA).
In vivo anti-tumor activity in H22-tumor-bearing mice
The H22 cell line (murine sarcoma) was obtained from the Chinese Academy of Medical Sciences & Peking Union Medical College Institute of Animals (Beijing, China).
The H22 cells (5 × 106 cells per mouse) were inoculated subcutaneously into the ICR mice in the axillary region. The H22-tumor-bearing mice were randomly divided into five groups (eight mice per group) and were injected via the lateral tail vein one time per day for 9 d after the second day of the tumor cell inoculation with one of the following treatments: BSA solution (100 mg/kg) (group 1), cyclophosphamide solution (50 mg/kg) (group 2), and PPD nanosuspensions of 20 mg/kg (group 3), 50 mg/kg (group 4), and 100 mg/kg (group 5). The mice were dissected, and the tumors were excised and weighed. The tumor inhibition rate was calculated using the following formula:
Statistical analysis
Pharmacokinetic parameters were calculated using non-compartmental methods with WinNonlin (V 4.0, Pharsight Corporation, St. Louis, MO). Statistical analysis of the inhibitory effect on tumor growth and bioavailability was performed using one-way analysis of variance. For p-values that were 0.05 or less, the difference was considered significant.
Results and discussion
Preparation of nanosuspensions
In preliminary experiments, several methods were examined for the preparation of PPD nanosuspensions, but the precipitation–ultrasonication method proved to be the most successful due to its good reproducibility.
Ideal nanosuspensions should meet the following requirements: (1) small size (about 200 nm) with a narrow size distribution; (2) stability in normal saline, PBS, rat plasma, or gastrointestinal fluids; (3) the absence of hemolysis; (4) facile reconstitution after lyophilization (reconstituted nanosuspensions showing no significant change in size or shape). For these purpose, a suitable stabilizer has to be selected from a series of excipients for the preparation of PPD nanosuspensions. The in vitro test demonstrated that PPD nanosuspensions prepared using BSA met all the above requirements. BSA has been widely used to prepare drug nanoparticles. Bovine serum albumin has a plurality of hydrophobic binding sites on surface, which can communicate with a variety of compounds, especially neutral or negatively charged with hydrophobic compounds. Structural domains of IIA and IIIA are considered to be of the two most critical binding sites. While PPD is a kind of water insoluble drug and the plasma protein-binding rate was 100%, therefore, which is suitable for preparing nanosuspension with bovine serum albumin as carrier. How BSA stabilizes nanoparticles has been reported and reviewed (Elzoghby et al., Citation2012).
Characterizations of nanosuspensions
Lyophilization
The newly prepared PPD nanosuspensions were stable under room temperature over 7 d (230 nm). To further enhance the storage stability, the freshly prepared PPD nanosuspensions (approximately 5 mg/ml) were lyophilized. BSA could perform as a cryoprotectant for lyophilization. PPD nanosuspensions could be easily lyophilized and then reconstituted with physiological saline to obtain nanosuspensions of similar size. The drug payload achieved 50% after lyophilization and the maximum PPD concentration can reach 100 mg/ml.
Sizes and zeta potentials
The mean particle size of the reconstituted PPD nanosuspensions was 222 ± 12 nm (n = 3), with the small polydispersity index being 0.118 ± 0.034 and zeta potential being−28.0 ± 0.5 mV (n = 3). The small polydispersity index value indicated a narrow size distribution. The particle size distribution is shown in .
Figure 1. The particle size distribution of nanosuspension (n = 3).
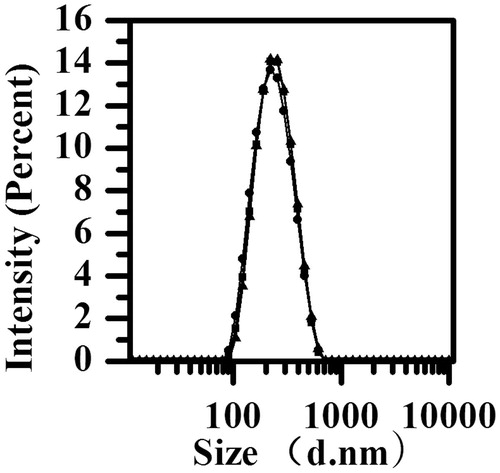
The zeta potential played an important effect on the storage stability of the colloidal dispersion system, and it reflected the electrostatic barriers that could prevent the nanoparticles to form aggregation and agglomeration that caused by low zeta potential. In general, a zeta potential of at least −30 mV for electrostatically stabilized systems or −20 mV for sterically stabilized systems is needed to obtain a physically stable nanosuspension (Jacobs et al., Citation2000). The zeta potential in the study could predict the storage stability of the nanosuspensions.
Morphology of nanosuspensions
The SEM observation revealed that the PPD nanosuspensions were nearly spherical, with smooth surfaces and approximately 210 nm in size (). In comparison, bulk PPD powder was much larger (over 2 μm), with irregular shapes, and composed mostly of fragmented drug crystals (). The size determined by SEM (210 nm) was smaller than that DLS (222 nm) because of the shrinkage of the PPD nanoparticles during SEM sample preparation (Zhang et al., Citation2007). In other words, the DLS determines the hydrodynamic diameter or the “equivalent sphere diameter” in solution, while SEM images were obtained in the absence of the solvent (Morita et al., Citation2001; Ye et al., Citation2002).
Figure 2. The SEM image of bulk PPD powder and PPD nanosuspensions ((a) PPD powder (× 9000), (b) PPD nanosuspensions (× 9000), and (c) PPD nanosuspensions (× 25 000)).

XRD and DSC
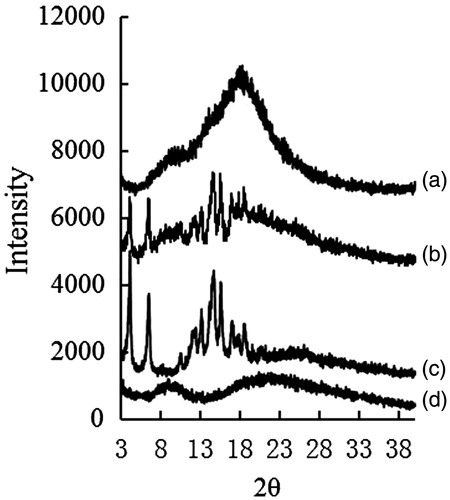
shows the patterns for the XRD spectra of bulk PPD powder, BSA, lyophilized PPD nanosuspensions, physical mixture of bulk PPD power, and BSA. The diffraction patterns of bulk PPD powder showed characteristic high-energy diffraction peaks at 2θ of 4.12°, 6.52°, and 14.68°, indicating a crystalline structure for PPD. The lyophilized PPD nanosuspensions displayed very flat curves with no intense peaks. These showed that the crystalline structure of PPD had been lost during the preparation process of the PPD nanosuspensions. Possibly, PPD was present in an amorphous state in the lyophilized PPD nanosuspensions.
Figure 3. The XRD of PPD nanosuspensions (a), physical mixture of bulk PPD power with BSA (b), bulk PPD power (c), and BSA (d).
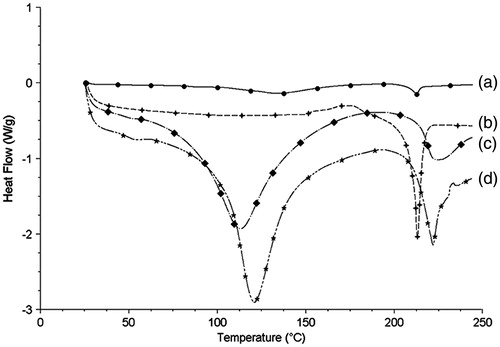
The DSC investigation () yielded similar results to those of the XRD study. Bulk PPD powder exhibited a sharp melting process with an endothermic peak at 212.5 °C. BSA exhibited a sharp melting process with an endothermic peak at 110 °C and 220 °C. No melting process was observed for the lyophilized PPD nanosuspensions, indicating that there was substantial conversion from the crystalline state to an amorphous state in PPD nanosuspensions during preparation.
Figure 4. The DSC thermograms of PPD nanosuspensions (a), bulk PPD power (b), BSA (c), and physical mixture of bulk PPD power with BSA (d).
In vitro cytotoxicity of nanosuspensions against HepG-2 cells
The cells were cultured with the PPD nanosuspensions and solution at concentrations of 0.625, 1.25, 2.5, 5, and 10 µg/ml for 12, 24, and 36 h. The results showed that nanosuspensions and solution significantly inhibit HepG2 cell growth and both showed the concentration- and time-dependent manner ().
Figure 5. The cytotoxicity of PPD nanosuspensions and PPD solution in the HepG2 cells after 12 h (a), 24 h (b), and 36 h (c) exposure. Results are expressed as mean ± SD (n = 6). *p < 0.05 and **p < 0.01 (nanosuspensions versus solution).
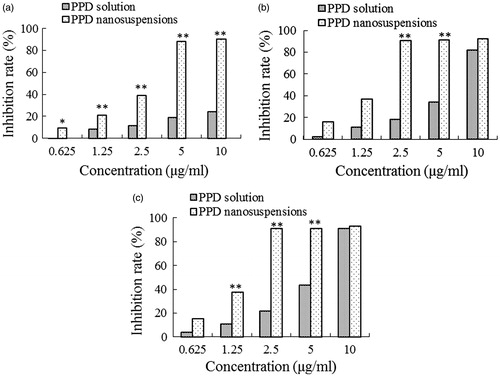
A significant increase in the inhibition rates was shown in the PPD nanosuspension-treated cells compared with the solution-treated cells under identical experimental conditions. The PPD nanosuspensions presented the stronger growth inhibition against HepG2 cells after longer incubation. In comparison with PPD solution, at 12 h, 10 µg/ml of PPD nanosuspensions achieved about 90% inhibition against HepG2 cell while PPD solution only 30%, the IC50 value of PPD nanosuspensions was apparently lower after the identical incubation time, accounting for 1.40 versus 5.83 µg/ml at 24 h and 1.39 versus 4.89 µg/ml at 36 h, respectively. These data suggested that PPD nanosuspensions were much more effective at inhibiting the growth of the HepG2 cells compared with the PPD solution. MTT assay demonstrated that the PPD nanosuspensions had a remarkable inhibitory effect on the proliferation of the HepG 2 cells. The inhibitory effect was increased with the concentration and incubation time. It was possible that, as other nanoparticle drug-delivery systems, the small size of the nanosuspension facilitated its adhesion to the cells and increased the contact area and time between the drug and the cells (Du et al., Citation2005; Lamprecht, Citation2010). In addition, BSA can also enhance the uptake of the drug into the tumor cells (Yardley, Citation2013). It is generally considered that nanoparticles can be nonspecifically internalized into cells via endocytosis or phagocytosis (Lou et al., Citation2011). Nanoparticles can also be adsorbed non-specifically by pinocytosis after accumulating on the surface of the cells. The above phenomena, as well as adhesion property, may facilitate the uptake of PPD nanosuspensions by tumor cells (Lou et al., Citation2011).
Pharmacokinetics and oral bioavailability studies
shows the mean plasma concentration–time curves in rats after the i.g. administration of 25 mg/kg of body weight of the PPD coarse suspensions and nanosuspensions. The PPD nanosuspensions maintained a high plasma drug concentration over a much longer period of time, in contrast to PPD coarse suspensions.
Figure 6. The mean plasma concentration–time curves of PPD in rats after the i.g. administration of the coarse suspensions and nanosuspensions at a single dose of 25 mg/kg of body weight (a) and the i.v. administration of the solution and nanosuspensions at a single dose of 25 mg/kg of body weight (b). Each point with a bar represents the mean ± SD (n = 6).
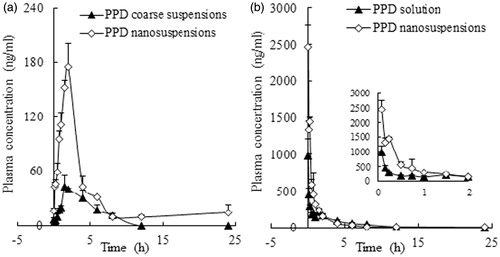
As shown in , when administered i.g., the Cmax and AUClast values of the PPD nanosuspensions were approximately 3.66-fold and 3.48-fold those of the PPD coarse suspensions. These finding indicated that PPD nanosuspensions in our study were more effective than previously reported (Jin et al., Citation2013c) in improving the PPD oral bioavailability, the relative bioavailability increased by 2.48-fold in comparison with bulk PPD powder, and the absolute bioavailability reached 66% in comparison with intravenous injected PPD solution. The improved oral bioavailability of the PPD nanosuspensions was explained as follows: first, the smaller particle size increased the surface area and reduced the thickness of the diffusion layer of the PPD nanosuspensions against the bulk PPD powder, leading to enhanced PPD dissolution and then enhanced absorption (Hintz & Johnson, Citation1989; Jia et al., Citation2002). Second, the adhesion between the nanoparticles and the intestinal villi prolongs the contact of the PPD nanosuspensions with the absorbing membranes of the gut (Lamprecht et al., Citation2006; Li et al., Citation2009). Third, the enhanced bioavailability could also be a result of the nanosuspension existing as amorphous drug compared with the coarse suspension existing as crystalline drug (Andeep et al., Citation2012). Finally, a small portion of the PPD nanosuspensions might pass through Peyer’s patch of the small intestine and be directly absorbed in nanoparticle form, as previously reported (Grama et al., Citation2011). Adhesion of the PPD nanosuspensions to intestinal villi could prolong the effective drug absorption time, which might be the reason that the PPD nanosuspensions showed an extended plasma MRT in comparison with the PPD coarse suspensions (4.41 h versus 3.33 h) and a longer t1/2 (9.42 h versus 2.99 h) after the oral administration. This finding indicated that the PPD nanosuspensions in our study were more effective in improving the oral bioavailability of PPD.
Table 1. Summary of the pharmacokinetic parameters after i.g. administration of two PPD formulations at a dose of 25 mg/kg in rats.
shows the mean plasma concentration–time curves in rats after i.v. administration of 25 mg/kg of body weight of the PPD solution and nanosuspensions. The PPD nanosuspensions maintained a higher plasma drug concentration than the solution, especially during the initial 2 h.
As shown in , the Cmax and AUClast values of the PPD nanosuspensions were approximately 2.8-fold and 1.62-fold those of the solution, respectively. The CI value of the PPD nanosuspensions was less than that of the solution (12 495.97 ml/h/kg versus 18 205.49 ml/h/kg), and the MRT of the PPD nanosuspensions was 1.5-fold that of the solution (3.16 versus 2.16 h). These findings revealed that if i.v. administrated, PPD nanosuspensions may have better therapeutic efficacy than PPD solution due to their prolonged circulation time and increased plasma drug level.
Table 2. Summary of the pharmacokinetic parameters after i.v. administration of two PPD formulations at a dose of 25 mg/kg of body weight in rats.
In vivo anti-tumor activity in H22-tumor-bearing mice
The in vivo anti-tumor effect of PPD nanosuspensions and cyclophosphamide solution was assessed using H22-tumor-bearing mice as the animal model. Cyclophosphamide is one of the most frequently used anti-tumor agents for the treatment of a broad spectrum of cancers, as a positive control in the study (Wang et al, Citation2007). Three doses of PPD nanosuspensions employed were to determine whether the PPD nanosuspensions inhibited effect on tumor in a concentration-dependent manner. These results showed that PPD nanosuspensions had a measurable inhibition effect on tumor growth at either a low dose or a high dose. The inhibition rate of 100 mg/kg of PPD nanosuspensions was 79.47%, while that of 50-mg/kg cyclophosphamide solution was 87.81%. A dose of 50 mg/kg or 20 mg/kg of PPD nanosuspensions had significant anti-tumor effects (the inhibition rates were 67.46% and 34.55%, respectively). These findings provide strong evidence that PPD nanosuspensions were a suitable drug delivery system for anti-tumor therapy.
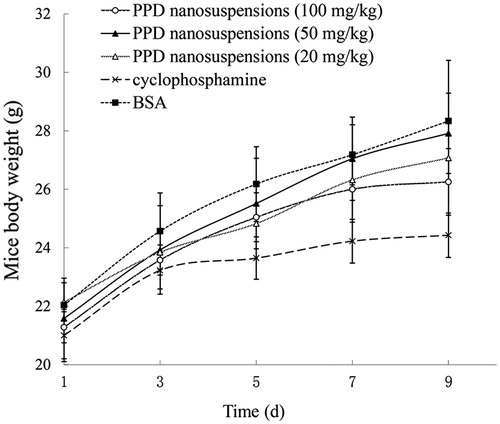
There was no significant difference (p > 0.05) in the mean body weight between the PPD nanosuspensions groups and the BSA group, whereas the body weight for the cyclophosphamide group increased much slower, suggested that the PPD nanosuspensions might have lower toxicity than cyclophosphamide. The body weight change is shown in .
Figure 7. The body weight changes in mice. Each value represents the mean ± SD (n = 8).
Conclusion
In our study, a precipitation–ultrasonication method was successfully employed to produce stable PPD nanosuspensions with a small size and a narrow size distribution. The drug payload achieved 50%, and no cryoprotectant was required for lyophilization. The drug in suspension is at a maximum concentration of unprecedented 100 mg/ml. The administration of the PPD nanosuspensions greatly increased the absolute oral bioavailability of PPD to 66%, which is the highest level reported so far. In comparison with the PPD free drug, the PPD nanosuspensions significantly improved the anti-tumor activity (p < 0.01) and achieved a good in vivo therapeutic effect. The nanosuspensions in this study could be used as an effective strategy for PPD delivery and improved therapeutic efficacy and might be used for other relatively insoluble drugs.
Declaration of interest
The authors are thankful to the National Natural Science Foundation of China for financial support (Project no. 81102813), Open Project for Key Laboratory, Heilongjiang University of Traditional Chinese Medicine (2013kf05) and the National S&T Major Special Project on Major New Drug Innovation (2012ZX09301002-001). The authors report that there are no conflicts of interest.
Related Research Data
References
- Andeep SC, Madgulkar AR, Kshirsagar SJ, et al. (2012). Amorphous nanoparticles for solubility enhancement. J Adv Pharm Sci 2:167–78
- Dong H, Bai LP, Wong KW, et al. (2011). The in vitro structure-related anti-cancer activity of ginsenosides and their derivatives. Molecules 16:10619–30
- Du D, Liu S, Chen J, et al. (2005). Colloidal gold nanoparticle modified carbon paste interface for studies of tumor cell adhesion and viability. Biomaterials 26:6487–95
- Elzoghby AO, Samy WM, Elgindy NA. (2012). Albumin-based nanoparticles as potential controlled release drug delivery systems. J Control Release 157:168–82
- Gao L, Zhang D, Chen MH, et al. (2008). Drug nanocrystals for the formulation of poorly soluble drugs and its application as a potential drug delivery system. J Nanopart Res 10:845–62
- Grama CN, Ankola DD, Ravi Kumar MNV. (2011). Poly (lactide-co-glycolide) nano-particles for peroral delivery of bioactives. Curr Opin Colloid Interface Sci 16:238–45
- Han MH, Chen J, Chen SL, et al. (2010). Development of a UPLC-ESI-MS/MS assay for 20(S)-protopanaxadiol and pharmacokinetic application of its two formulations in rats. Anal Sci 26: 749–53
- Han MH, Chen J, Wang YQ, et al. (2013). Determination of 20(S)-protopanaxadiol in rat plasma by LC-MS/MS method and its application to the pharmacokinetic study: a comparative study of its solution and two oral formulations. J Anal Chem 68:1–6
- Hasegawa H, Lee KS, Nagaoka T, et al. (2000). Pharmacokinetics of ginsenoside deglycosylated by intestinal bacteria and its transformation to biologically active fatly acid esters. Biol Pharm Bull 23:298–304
- Hintz RJ, Johnson KC. (1989). The effect of particle size distribution on dissolution rate and oral absorption. Int J Pharm 51:9–17
- Iishi H, Tatsuta M, Baba M, et al. (1997). Inhibition by ginsenoside Rg3 of bombesin-enhanced peritoneal metastasis of intestinal adenocarcinomas induced by azoxymethane in Wistar rats. Clin Exp Metastasis 15:603–11
- Jacobs C, Kayser O, Muller RH. (2000). Nanosuspensions as a new approach for the formulation for the poorly soluble drug tarazepide. Int J Pharm 196:161–4
- Jia L, Wong HC, Cerna S, et al. (2002). Effect of nanonization on absorption of 301029: ex vivo and in vivo pharmacokinetic correlations determined by liquid chromatography/mass spectrometry. Pharm Res 19:1091–6
- Jin X, Zhang ZH, Li SL, et al. (2013a). A nanostructured liquid crystalline formulation of 20(S)-protopanaxadiol with improved oral absorption. Fitoterapia 84:64–71
- Jin X, Zhang ZH, Sun E, et al. (2013b). Study on pharmacokinetics of 20 (S)-protopanaxadiol lipid cubic nanoparticles. Zhongguo Zhong Yao Za Zhi 38:263–8
- Jin X, Zhang ZH, Sun E, et al. (2013c). Enhanced oral absorption of 20(S)-protopanaxadiol by self-assembled liquid crystalline nanoparticles containing piperine: in vitro and in vivo studies. Int J Nanomed 8:641–52
- Keck CM, Müller RH. (2006). Drug nanocrystals of poorly soluble drugs produced by high pressure homogenization. Eur Pharm Biopharm 62:3–16
- Lamprecht A. (2010). IBD: selective nanoparticle adhesion can enhance colitis therapy. Nat Rev Gastroenterol Hepatol 7:311–12
- Lamprecht A, Koening P, Ubrich N, et al. (2006). Low molecular weight heparin nanoparticles: mucoadhesion and behaviour in Caco-2 cells. Nanotechnology 17:3673–80
- Lee SJ, Ko WG, Kim JH, et al. (2000). Introduction of apoptosis by a novel intestinal metabolite of ginseng saponin via cytochrome c-mediated activation of caspase-3 protease. Biochem Pharmacol 60:677–85
- Li G, Wang Z, Sun Y, et al. (2006). Ginsenoside 20(S)-protopanaxadiol inhibits the proliferation and invasion of human fibrosarcoma HT1080 cells. Basic Clin Pharmacol Toxicol 98:588–92
- Li L, Chen X, Li D, et al. (2011). Identification of 20(S)-protopanaxadiol metabolites in human liver microsomes and human hepatocytes. Drug Metab Dispos 39:472–83
- Li W, Liu Y, Zhang JW, et al. (2009). Anti-androgen-independent prostate cancer effects of ginsenoside metabolites in vitro: mechanism and possible structure–activity relationship investigation. Arch Pharm Res 32:49–57
- Li XM, Gu L, Xu YL, et al. (2009). Preparation of fenofibrate nanosuspension and study of its pharmacokinetic behavior in rats. Drug Dev Ind Pharm 35:827–33
- Liu GY, Bu X, Yan H, et al. (2007). 20(S)-protopanaxadiol-induced programmed cell death in glioma cells through caspase-dependent and -independent pathways. J Nat Prod 70:259–64
- Lou HY, Gao L, Wei XB, et al. (2011). Oridonin nanosuspension enhances anti-tumor efficacy in SMMC-7721 cells and H22 tumor bearing mice. Colloids Surf B: Biointerfaces 87:319–25
- Morita T, Horikiri Y, Suzuki T, et al. (2001). Preparation of gelatin microparticles by co-lyophilization with poly (ethylene glycol): characterization and application to entrapment into biodegradable microspheres. Int J Pharm 219:127–37
- Ota T, Maeda AM, Odashima S. (1991). Mechanism of action of ginsenoside Rh2: uptake and metabolism of ginsenoside Rh2 by cultured B16 melanoma cells. J Pharm Sci 80:1141–6
- Popovich DG, Kitts DD. (2002). Structure–function relationship exists for ginsenosides in reducing cell proliferation and inducing apoptosis in the human leukemia (THP-1) cell line. Arch Biochem Biophys 406:1–8
- Popovich DG, Kitts DD. (2004). Ginsenosides 20(S)-protopanaxadiol and Rh2 reduce cell proliferation and increase sub-G1 cells in two cultured intestinal cell lines, Int-407 and Caco-2. Can J Physiol Pharmacol 82:183–90
- Usami Y, Liu YN, Lin AS, et al. (2008). Antitumor agents. 20(S)-protopanaxadiol and 20(S)-protopanaxatriol as antiangiogenic agents and total assignment of 1H NMR spectra. J Nat Prod 71:478–81
- Wang W, Zhao YQ, Rayburn ER, et al. (2007). In vitro anti-cancer activity and structure–activity relationships of natural products isolated from fruits of Panax ginseng. Cancer Chemother Pharmacol 59:589–601
- Wang X, Zhang J, Xu T. (2007). Cyclophosphamide as a potent inhibitor of tumor thioredoxin reductase in vivo. Toxicol Appl Pharmacol 218:88–95
- Xia HJ, Zhang ZH, Jin X, et al. (2013). A novel drug–phospholipid complex enriched with micelles: preparation and evaluation in vitro and in vivo. Int J Nanomed 8:545–54
- Yardley DA. (2013). nab-Paclitaxel mechanisms of action and delivery. J Control Release 170:365–72
- Ye Q, Zhang ZC, Jia HT, et al. (2002). Formation of monodisperse polyacrylamide particles by radiation-induced dispersion polymerization: particle size and size distribution. J Colloid Interface Sci 253:279–84
- Yu Y, Zhou O, Hang Y, et al. (2007). Antiestrogenic effect of 20(S)-protopanaxadiol and its synergy with tamoxifen on breast cancer cells. Cancer 109:2374–82
- Zhang LY, Yang M, Wang Q, et al. (2007). 10-Hydroxycamptothecin loaded nanoparticles: preparation and antitumor activity in mice. J Contorl Release 119:153–62
- Zhu GY, Li YW, Tse AKW, et al. (2011). 20(S)-protopanaxadiol, a metabolite of ginsenosides, induced cell apoptosis through endoplasmic reticulum stress in human hepatocarcinoma HepG2 cells. Eur J Pharmacol 668:88–98