
Abstract
Darunavir is effective against wild-type and PI-resistant HIV, and has an oral bioavailability of 37%. It needs to be combined with ritonavir, which increases the bioavailability to 82%. The aim of this study was to evaluate the in-vivo efficacy of the darunavir-SLN and demonstrate lymphatic transport as a contributing pathway in increasing the drug bioavailability. The SLN was prepared by hot-homogenization technique using GMS as lipid. In-vitro drug release from SLN at the 12th hour was retarded (80.6%) compared to marketed tablet (92.6%). Ex-vivo apparent permeability of the freeze-dried SLN across everted rat intestine was 24 × 10−6 at 37 °C and 5.6 × 10−6 at 4 °C. The presence of endocytic process inhibitors like chlorpromazine and nystatin reduced it to 18.8 × 10−6 and 20.2 × 10−6, respectively, which established involvement of endocytic mechanism in the uptake of SLN. In-vivo pharmacokinetic studies on rats demonstrated increase in the AUC of SLN (26) as compared to that of marketed tablet (13.22), while the presence of lymphatic uptake inhibitor cycloheximide lowered the AUC of SLN to 17.19 which further led credence to the involvement of lymphatic uptake behind improved bioavailability. The detection of darunavir in the lymphatic fluid of the rats administered with darunavir-SLN further reinforced the conclusion of SLN being taken up by the lymphatic system.
Introduction
Oral route of administration has always been favored owing to the ease in manufacturing and patient compliance. The oral bioavailability of anti retrovirals has been a challenging topic in recent years due to drug properties such as low-aqueous solubility, substrate for CYP-mediated metabolism, substrate for P-gp efflux, etc., which reduce the bioavailability to an enormous extent (Vermeir et al., Citation2009). The captivation of such drugs from the gastrointestinal tract is a multifarious process and is poorly characterized affecting the range of the systemic drug exposure levels (Charman & Stella, Citation1991). The approaches which are being exploited to overcome these limitations include chemical modification of pre-existing entities, designing of new chemically modified entity, close examination of various dosing regimens and development of novel delivery systems to improve the efficacy of both prevailing as well as newly developed anti-retroviral drugs. Lymphatic system is found to play a major role in the oral absorption of lipids and lipophilic drugs from intestine, and thus, it can be used as a pathway for bioavailability enhancement of drugs which are prone to hepatic metabolism (Reddy & Murthy, Citation2002).
Darunavir, a non-peptidic protease inhibitor, suffers from poor oral bioavailability (37%) owing to it being a substrate for cyp3A metabolism and also to the poly glycoprotein system (PgP) which causes efflux of the absorbed drug back into the intestinal lumen. The bioavailability of darunavir can be increased to 82% using ritonavir, a potent cyp3A inhibitor, but the same increases additional drug load on the patients and also affects the total dosage cost. Despite of all these, low solubility of darunavir in water (0.15 mg/ml), in lipids (log p 1.8) and the degradation above melting point (74 °C) render its formulation as solid lipid nanoparticles (SLN) a challenge.
The objective of this study was to develop a novel ritonavir-independent method for the improvement of oral bioavailability of darunavir. The approach used to overcome the hitches relating to formulation is to incorporate darunavir in SLN which are demonstrated herein to bypass Cyp3A metabolism in the liver owing to use of lymphatic route transportation and thus there is an increase in the oral bioavailability (Reddy & Murthy, Citation2002).
Materials and methods
Materials
Darunavir was received as a kind gift from Lupin Research Park, Pune, India, and all other chemicals were procured from the local sources.
Methods
Preparation and evaluation of darunavir-SLN
Glyceryl caprylate (1 ml) was used as a co-solubilizer to dissolve darunavir (500 mg) in glyceryl monosterate (GMS) (1.5 g) maintained at 60 °C to which Span 80 (1 g) was added. Aqueous phase (2% Tween 80 solution in distilled water) also maintained at 60 °C and was added to the above lipid phase and emulsified at 15 000 rpm using an overhead stirrer (Remi Equipments, Mumbai, India); the resultant emulsion was passed through Avestin Emulsiflex (Ottawa, Canada) C3 high-pressure homogenizer (HPH) at 500 bars (1 cycle). Further, the SLN dispersion was lyophilized using aerosil (100% w/w of drug) as cryoprotectant. The obtained SLN was evaluated for particle size (PS) and zeta potential using Malvern Zetasizer ZS 90, the morphology of SLN was determined by transmission electron microscopy (TEM) (FEI Corporation, Gräfelfing, Germany), and the entrapment efficiency (EE) was determined by dissolving fixed amount of lyophilized SLN in chloroform and determining the drug content spectrophotometrically at 262 nm. Micromeritic properties such as angle of repose, bulk density and Carr index were determined using methods reported in literature (Lachman & Lieberman, Citation2009).
In-vitro release of darunavir in simulated gastric condition
The freeze-dried SLN was subjected to in-vitro drug release study in USP dissolution apparatus (Labindia Analytical) Type II using dissolution medium 0.1N HCl for 2 h and further in phosphate buffer solution pH 6.8, thus mimicking the GIT condition. The change in pH of the medium was achieved using pH change method involving addition of sodium phosphate tribasic dodecahydrate (Goyal & Vashist, Citation2011). The sampled aliquots were analyzed spectrophotometrically at 264 nm to determine the drug content. The kinetics of drug release was studied using PCP disso software. Regression coefficient and time for 50% drug release were calculated using the same.
Ex-vivo studies
Literatures explain numerous methods for determination of the uptake of lipid-based nano-formulations from intestine (Lind et al., Citation2008; Roger et al., Citation2010; Sahay et al., Citation2010). In order to establish the uptake mechanism of freeze-dried darunavir-SLN, everted gut sac studies for the same were performed on intestinal segments of male Wistar rats. For the uptake study, one end of the isolated intestine was clamped and secured with a silk suture, while from the other open end 1 ml of phosphate buffer of pH 6.8 was filled using a syringe. The proximal end was then carefully secured using silk suture, and the resultant sac was incubated in a dispersion of darunavir-SLN (effective concentration equivalent to 5 μg/ml).
To evaluate the uptake mechanism of the SLN by the intestinal cells in detail, everted gut sacs as described above were also incubated separately in darunavir-SLN dispersions at 4 °C & with specific endocytic inhibitors like chlorpromazine (CPZ) (10 µg/ml) and nystatin (NYT) (25 µg/ml) at 37 °c for 30 min. Post-incubation, intestinal sacs were carefully removed, and the contents were collected in test tubes. Sacs were rinsed thrice with KHB buffer, and rinsings were combined with original content for spectrophotometric analysis.
The analysis for quantification of the drug permeated across the intestine was done spectrophotometrically at 267 nm by diluting 1 ml of the above collected samples from everted intestine and diluting up to 10 ml with methanol, followed by sonication for 45 min.
In-vivo pharmacokinetic study
Permission for carrying out the in-vivo studies was obtained from Institutional Animal Ethics Committee (IAEC) of AISSMS College of Pharmacy, and their guidelines were followed during course of the study. Male Wistar rats weighing 180–250 g were used for the study. Two groups namely groups I and II containing six rats each were formed. Group I received darunavir marketed tablet (10 mg/kg of body weight), while group II received darunavir-SLN (10 mg/kg of body weight).
The animals were fasted overnight (12 h) prior to the dosing. Freeze-dried SLN and marketed darunavir tablet (crushed to powder) were suspended in distilled water using 0.5% w/v methyl cellulose and fed to the animals with the aid of oral feeding needle. Blood samples (2 m) were withdrawn from the retro-orbital plexus at 0, 2, 4, 6 and 8 h time interval and collected in EDTA-coated tubes. The blood samples were centrifuged immediately at 15 000 rpm for 10 min, and the plasma was separated which was stored at −20 °C in screw-capped polypropylene tubes till the time of analysis. The darunavir content in plasma samples was quantified by HPLC (Jasco Model PU 2080 Plus pump, Tokyo, Japan) using acetonitrile and water (1:1) as mobile phase at a flow rate of 1 ml/min.
Effect of lymphatic uptake blocker on the in-vivo uptake of SLN in rats
In order to study the uptake of the prepared darunavir-SLN by the intestinal enterocytes, in-vivo studies were designed in the presence and absence of cycloheximide (CXI), a well-known lymph transport inhibitor. CXI is known to inhibit the secretion of chylomicrons from the enterocytes and, therefore, inhibits the lymphatic transport of the lipidic entities without causing damage to other active and passive absorption pathways (Dahan & Hoffman, Citation2005; Lind et al., Citation2008; Gao et al., Citation2011; Zhang et al., Citation2012). For the above study, male Wistar rats weighing 180–250 g were used, and the same were divided into two groups, namely groups I and II.
The animals were fasted overnight (12 h) prior to dosing. Group I received CXI saline solution 1 h prior to dosing of darunavir-SLN (10 mg/kg of body weight), while group II received only darunavir-SLN (10 mg/kg of body weight). Feeding of darunavir-SLN, blood withdrawal and analysis were done in a similar manner as described in “In-vivo Pharmacokinetic study” section.
Detection of the drug concentration in lymph fluid of rat
In order to establish conclusive evidence of lymphatic uptake of darunavir-SLN, the lymphatic fluid of the treated rats was evaluated for darunavir content. Male Wistar rats (n = 6) weighing 180–250 g were chosen for the surgery. The rats were fed with suspension of freeze-dried SLN (equivalent to 10 mg/kg of darunavir) in 0.2% methyl cellulose solution in distilled water prior to surgery. Surgical procedure was carried out to locate the mesenteric lymphatic duct of the rat upon location of which, cannulation was done to withdraw lymph fluid (Wasan & Boyd, Citation2004). The darunavir content in lymphatic fluid was quantified by validated HPLC method.
Results and discussion
Preparation and evaluation of darunavir-SLN
The darunavir-SLN obtained by hot homogenization method had particle size of 210 nm and EE of 74.23%. Freeze-dried SLN were found to have PS and zeta potential of 270 nm and −22 mv, respectively, thus, owing to the small size the particles would be easily taken up in the lymphatic system, and the negative charge would prevent the SLN from entangling in the negatively charged mucous. The % entrapment of the freeze-dried SLN was found to be 69.8% and the same was found to have spherical shape and smooth morphology (). The excellent flow ability or free flowing nature depicted by angle of repose, bulk density and Carr index () were due to the spherical morphology and aerosil matrix; thus, improving formulation compliant to further processing into dosage form.
Figure 1. TEM image of freeze-dried SLN.
Table 1. Micromeritic properties of freeze-dried darunavir-SLN (n = 3).
In-vitro release of darunavir in simulated gastric condition
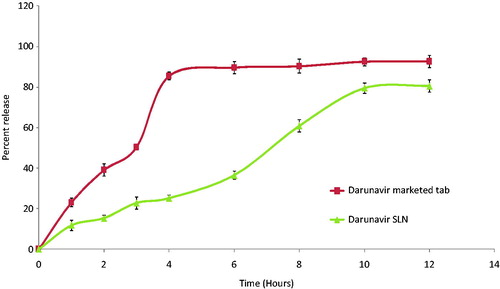
The drug release () from SLN in the simulated gastric condition was found to be sustained (80.6% in 12 h) and the same followed Peppas model with n = 0.998 and k = 7.349 resembling an anomalous release mechanism involving diffusion and erosion.
Figure 2. In vitro release profile of darunavir-SLN in simulated gastric condition (n = 6, mean ± SD).
Ex-vivo studies
Literature identifies numerous energy-dependent endocytic processes for the uptake of nanoparticles, principally clathrin-mediated endocytosis and caveolae-mediated endocytosis which are receptor-mediated processes and clathrin–caveolae-independent endocytic processes, importantly, macropinocytosis which are actin-dependent and non-receptor-mediated process (Roger et al., Citation2010; Sahay et al. Citation2010;). Through macropinocytosis particles smaller than 2 μm can be uptaken into enterocytes (Roger et al., Citation2010).
To explicate the uptake of SLN by the above-mentioned receptor-mediated process, the experiment was carried out in the presence of chemicals which are known inhibitors of clathrin- and caveolae-mediated endocytic processes. CPZ, a cationic amphiphilic drug, is reported to inhibit clathrin-mediated endocytosis by causing accumulation of clathrin in late endosomes and reducing its expression on the enterocytic surface (Sahay et al. Citation2010). Caveolae are special type of cholesterol and sphingo lipids-rich rafts, flask-shaped infolded structures on the plasma membrane to engulf cargo molecules or carriers binding to their surface. The same is blocked by the antifungal drug NYT because of cholesterol sequestration. Blocking of the endocytosis process is observed at low temperatures, hence one experiment was designed at 4 °C (Lind et al., Citation2008). Macropinocytosis being unspecific was felt unnecessary to evaluate.
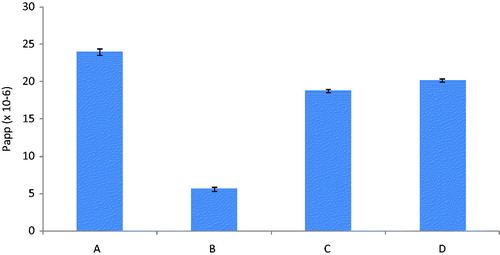
represents apparent permeability of the darunavir-SLN at 37 °C, 4 °C, in the presence of CPZ at 37 °C and in the presence of NYT at 37 °C measured in 30 min. It was evident from the results that the apparent permeability in the presence of CPZ was reduced by 21.6% as compared to that in the absence of CPZ, i.e. at 37 °C (taken to be as 100%). Similarly, the apparent permeability in the presence of NYT was also found to be reduced by 15.8% thus establishing the contribution of both clathrin- and caveolae-mediated process in the uptake of SLN. Also the reduction of the apparent permeability of the SLN at 4 °C by 76.6% as compared to that at 37 °C confirmed involvement of the mechanism as endocytic processes are totally ineffective at lower temperatures.
Figure 3. Ex-vivo apparent permeability of Dar SLN at (a) 37 °C, (b) 4 °C, in the presence of (c) CPZ at 37 °C and (d) NYT at 37 °C (n = 3).
In-vivo pharmacokinetic study
Evaluation of the in-vivo effectiveness of the freeze-dried SLN was executed by performing in-vivo pharmacokinetic study on male Wistar rats. The plasma drug concentration versus time profile is represented in .
Figure 4. In-vivo plasma concentrations profile of marketed tablet and darunavir-SLN in rats (n = 6, ± SD).
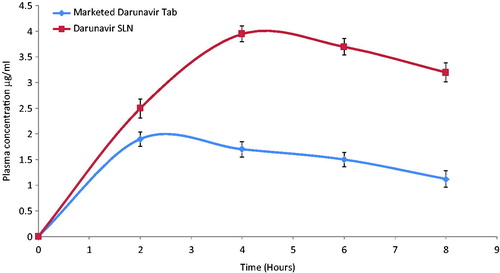
Compared to the marketed tablet, a significant increase in the AUC0−t, Cmax and Tmax was observed () for the darunavir-SLN. Apparently, it was concluded that incorporating darunavir in SLN helped to bypass the hepatic metabolism thereby increasing the Cmax and the AUC. It can be assumed that the SLN could have been adsorbed via the intestinal lymphatic region which ensured an increase in the AUC (Reddy & Murthy, Citation2002). Comparison of the various pharmacokinetic parameters of the marketed tablet and darunavir-SLN is summarized in .
Table 2. In-vivo pharmacokinetic parameters of marketed tablet and darunavir-SLN in rats.
Effect of lymphatic uptake blocker on the in-vivo uptake of SLN in rats
Uptake of the lipidic particles by the enterocytes of the intestine and transport to the systemic circulation via the lymphatic route has been reported in the literature (Jani et al., Citation1989). Primarily, there exists two mechanisms for the lipidic particles to get access to the lymphatic circulation; the first Peyer's patches including the gut associated lymphoid tissue (GALT) and lymphoid follicles and secondly triglyceridic lipoproteins (chylomicron assisted) (Khan & Mudassir, Citation2013). To evaluate, the lymphatic transport of darunavir-SLN pharmacokinetic study was performed on CXI-treated male Wistar rats. CXI, a protein synthesis inhibitor, is known to inhibit the lymphatic transport system without affecting the active and passive transports (Dahan & Hoffman, Citation2005; Lind et al., Citation2008).
It is evident from that CXI-treated rats showed lesser plasma concentration of darunavir as compared to that of the control group (CXI non-treated); this could be attributed to CXI-induced blockage of intestinal lymphatic transport. Comparison of the various pharmacokinetic parameters of CXI-treated and CXI-non-treated groups are summarized in .
Figure 5. In-vivo plasma concentrations profile of darunavir-SLN in CXI-treated and CXI-non-treated rats (n = 6, ± SD).
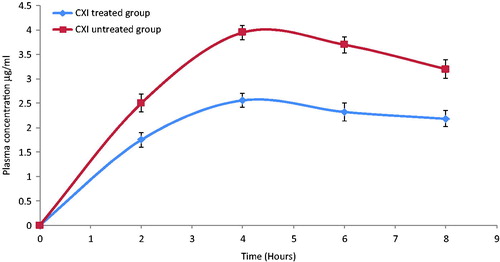
Table 3. In-vivo pharmacokinetic parameters of darunavir-SLN in rats in the presence and absence of lymphatic uptake blockers (CXI).
Detection of the drug concentration in lymph fluid of rat
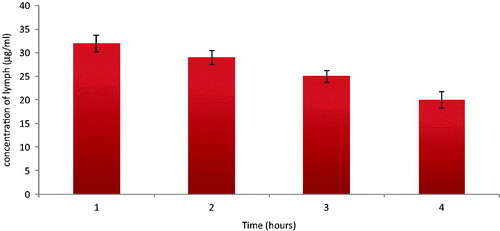
It is evident from the literature that the intestinal enterocytes, triglyceride-rich lipoproteins mediated transport and payers patch transport the lipidic particles to the lymphatic circulation and ultimately to the systemic circulation. As discussed in “Effect of lymphatic uptake blocker on the in-vivo uptake of SLN in rats” section, the uptake of lipidic particles is primary through the intestinal lymphatic route. The particles taken up from the intestine transport through the lymphatic capillaries to lymphatic collecting vessel from which it further passes to the lymphatic ducts via the lymph trunks. In order to evaluate the transport of SLN via the lymphatic route, mesenteric lymphatic duct of male Wistar rats was cannulated, and lymph was collected and analyzed for darunavir content.
The concentrations of darunavir in the lymph fluid found at 1st, 2nd, 3rd and 4th hour () conclusively establish the uptake and transport of darunavir-SLN to the systemic circulation via lymphatic route.
Figure 6. Concentration of darunavir in the lymphatic fluid of rats (n = 6, ± SD).
Conclusion
The role of lymphatic route in enhancing the oral bioavailability of darunavir was successfully demonstrated from the ex-vivo endocytic uptake studies and in-vivo studies in the presence and absence of lymphatic uptake inhibitor. The presence of drug in the lymphatic circulation evidently signposted the involvement of lymphatic route in the enhancement of the availability of the drug in the systemic circulation which was reflected by the increased AUC values from SLN over that from marketed tablets. Thus, it can be concluded that darunavir-SLN can be a potential drug delivery vehicle for improving oral bioavailability of darunavir abating the need to co-administer ritonavir and thus improving patient compliance and reduction in cost of therapy.
Declaration of interest
The authors have no interest to declare.
References
- Vermeir M, Lachau-Durand S, Mannens G. (2009). Absorption, metabolism, and excretion of darunavir: a new protease inhibitor administered alone and with low-dose ritonavir in healthy subjects. Drug Metab Dispos 37:809–20
- Charman WN, Stella VJ. (1991). Transport of lipophilic molecules by the intestinal lymphatic system. Adv Drug Deliv Rev 7:1–14
- Reddy LH, Murthy RSR. (2002). Lymphatic transport of orally administered drug. Indian J Exp Biol 40:1097–109
- Lachman L, Lieberman HA. (2009). The theory and practice of industrial pharmacy. New Delhi, India: CBS Publishers and Distributors, 313–17
- Goyal S, Vashist H. (2011). Development of alginate gel beads-entrapped liposome for colon specific drug delivery of prednisolone. Der Pharm Sin 2:31–8
- Sahay G, Alakhova D, Kabanov A. (2010). Endocytosis of nanomedicines. J Control Release 145:182–95
- Roger E, Lagarce F, Garcion E, Benoit JP. (2010). Biopharmaceutical parameters to consider in order to alter the fate of nanocarriers after oral delivery. Nanomedicine (London) 5:287–306
- Lind M, Jacobsen J, Holm R, Müllertz A. (2008). Intestinal lymphatic transport of halofantrine in rats assessed using a chylomicron flow blocking approach: the influence of polysorbate 60 and 80. Eur J Pharm Sci 35:211–18
- Dahan A, Hoffman A. (2005). Evaluation of a chylomicron flow blocking approach to investigate the intestinal lymphatic transport of lipophilic drugs. Eur J Pharm Sci 24:381–8
- Gao F, Zhang Z, Bu H, et al. (2011). Nanoemulsion improves the oral absorption of candesartan cilexetil in rats: performance and mechanism. J Control Release 149:168–74
- Zhang Z, Gao F, Bu H, et al. (2012). Solid lipid nanoparticles loading candesartan cilexetil enhance oral bioavailability: in vitro characteristics and absorption mechanism in rats. Nanomedicine 8:740–7
- Wasan K, Boyd M. (2004). A stepwise surgical procedure to investigate the lymphatic transport of lipid-based oral drug formulations: cannulation of the mesenteric and thoracic lymph ducts within the rat. J Pharmacol Toxicol 49:115–20
- Jani P, Halbert GW, Langridge J, Florence AT. (1989). The uptake and translocation of latex nanospheres and microis spheres after oral administration to rats. J Pharm Pharmacol 41:809–12
- Khan AA, Mudassir J. (2013). Advanced drug delivery to the lymphatic system: lipid based nanoformulations. Int J Nanomed 8:2733–44