
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
P-glycoprotein (P-gp) efflux is the major cause of multidrug resistance (MDR) in tumors when using anticancer drugs, moreover, poor bioavailability of few drugs is also due to P-gp efflux in the gut. Rapamycin (RPM) is in the clinical trials for breast cancer treatment, but its P-gp substrate property leads to poor oral bioavailability and efficacy. The objective of this study is to formulate and evaluate nanoparticles of RPM, along with a chemosensitizer (piperine, PIP) for improved oral bioavailability and efficacy. Poly(d,l-lactide-co-glycolide) (PLGA) was selected as polymer as it has moderate MDR reversal activity, which may provide additional benefits. The nanoprecipitation method was used to prepare PLGA nanoparticles with particle size below 150 nm, loaded with both drugs (RPM and PIP). Prepared nanoparticles showed sustained in vitro drug release for weeks, with initial release kinetics of zero order with non-Fickian transport, subsequently followed by Higuchi kinetics with Fickian diffusion. An everted gut sac method was used to study the effect of P-gp efflux on drug transport. This reveals that the uptake of the RPM (P-gp substrate) has been increased in the presence of chemosensitizer. Pharmacokinetic studies showed better absorption profile of RPM from polymeric nanoparticles compared to its suspension counterpart and improved bioavailability of 4.8-folds in combination with a chemosensitizer. An in vitro cell line study indicates higher efficacy of nanoparticles compared to free drug solution. Results suggest that the use of a combination of PIP with RPM nanoparticles would be a promising approach in the treatment of breast cancer.
Introduction
Breast cancer is one of the leading causes of cancer deaths in the world. Multidrug resistance (MDR) is a condition whereby cancer cells become resistant to the cytotoxic effects of various structurally and mechanistically unrelated chemotherapeutic agents. This is a major problem in the clinical treatment of cancer (Ho et al., Citation2007). Rapamycin (RPM) is a macrolide obtained from bacterium Streptomyces hygroscopicus. RPM has proven to be a versatile compound with several seemingly unrelated properties, including antifungal, immunosuppressive and anticancer. The “National Cancer Institute Developmental Therapeutics Program” demonstrated that RPM inhibits cell growth in tumor cell lines (Seto, Citation2012). This led to its development as a cancer therapeutic, either singly or in combination. RPM is now in the clinical trial phase for breast cancer (Seto, Citation2012; Abu-Khalaf, Citation2014). Although RPM promises high potency, its limited solubility of 2.6 μg/ml in water, low tumor specificity, dose dependent toxicity and low oral bioavailability limit its clinical use (Simamora et al., Citation2001). Like most of the anticancer drugs, RPM is effluxed out of tumor cells, mediated by P-glycoprotein (P-gp), MDR-associated protein 1 (MRP1), MRP2 and MRP3 (Ambudkar et al., Citation2003; Borst et al., Citation2006). The over expression of P-gp encoded by MDR1 gene is a major obstacle in cancer therapy. P-gp efflux can also be correlated to low oral bioavailability, since the drug is pumped back. To increase the cancer therapy effectiveness of anticancer drugs, the administration of anticancer drugs in combination with chemosensitizers for the treatment of MDR tumors has been investigated widely (Ganta & Amiji, Citation2009; Patil et al., Citation2009; Song et al., Citation2009).
Piperine (PIP) is a natural alkaloid obtained from Piper longum. It is well known for improving intestinal permeability (Shoba et al., Citation1998), as a P-gp inhibitor (Bhardwaj et al., Citation2002), and as an antioxidant (Vijayakumar et al., Citation2004) and it also exhibits mild anticancer activity (Bezerra et al., Citation2006). PIP also enhances clinical bioavailability in animal studies and trials on human volunteers (Shoba et al., Citation1998; Hoffman, Citation2013). This effect is attributed to its ability to enhance permeation and inhibit metabolizing enzymes, thereby circumventing first pass metabolism. The combination of PIP with anticancer drugs may provide the advantage of inhibiting P-gp efflux and improving drug accumulation in cancer cells and thus the anticancer efficacy of RPM will be enhanced. Moreover, the inherent activity of PIP against cancer cells gives the advantage of synergistic action against the tumor.
A nanoparticulate drug delivery system yields the advantage of controlled distribution and release of a drug in the body system. RPM nanoparticles showing such an effect has been reported (Das et al., Citation2008; Haddadi et al., Citation2008). As per author's knowledge, RPM has been reported as an individual nanoparticulate formulation but never reported with PIP. The current study involves biodegradable nanoparticles made from poly(d,l-lactide-co-glycolide) (PLGA), as drug carriers, PLGA nanoparticles possess moderate MDR reversal activity on their own (Sahoo & Labhasetwar, Citation2005), which may provide additional benefit with the drug and chemosensitizer. This forms the basis for selection of the class of drugs, the polymer and the delivery system. Thus, the present study deals with formulation, characterization, in vitro and in vivo evaluation of polymeric nanoparticles of RPM with PIP for oral delivery in cancer treatment.
Materials and methods
Materials
Piperine was generously donated by Indo Vedic Nutrients Pvt. Ltd., Bangalore, India. PLGA (50:50) 30–50 kDa, poly vinyl alcohol (PVA) and Vitamin E TPGS (d-alpha tocopheryl polyethylene glycol 1000 succinate) were purchased from Sigma-Aldrich (Bangalore, India). Tween 20 and Tween 80 were purchased from Lobachem (Mumbai, India). All the solvents used in high performance liquid chromatography (HPLC) were purchased from Merck Pvt Ltd. (Mumbai, India). Other chemicals used in the experiments were of analytical grade.
Preparation of nanoparticles
PLGA nanoparticles were prepared by the nanoprecipitation method as described by Chang et al. (Citation2009), with slight modifications. Briefly, 10 mg polymer was dissolved in 1 ml acetone and to this 1 mg RPM was added. This polymeric solution containing drug was added to the 10 ml surfactant solution (type and concentration of surfactant was varied in optimization) in aqueous phase during homogenization for 2 min. Then the formulation was stirred at 1000 rpm to facilitate the evaporation of organic solvent leaving behind the polymeric nanoparticles dispersed in aqueous phase. Prepared nanoparticles were centrifuged at 12 000rpm for 30 min to obtain nanoparticles pellets. Various parameters were optimized, including surfactant type, surfactant concentration and polymer concentration. Finally, the optimized nanoprecipitation method was used to prepare RPM- and PIP-loaded nanoparticles individually and also to prepare co-encapsulated nanoparticles containing both the drugs.
Particle size and size distribution
Particle size and size distribution were assessed using dynamic light scattering technique with zeta sizer (Nano-ZS, Malvern Instruments, Worcestershire, UK) and analyzed by “DTS Nano” software. All the formulations were properly diluted with millipore water and were shaken vigorously to get sufficient count rate. The average particle size and poly dispersity index (PDI) were recorded for all formulations.
Entrapment efficiency of nanoparticles
Entrapment efficiency was evaluated by analyzing the drug present in supernatant, which was obtained after centrifugation of nanoparticles at 12 000rpm for 30 min. Analysis was carried out for RPM by reverse phase HPLC (RP-HPLC) (Shimadzu UFLC model) on C-18 column Inertsil® (Octadecylsilane[ODS]-3 V, dimension 4.6 × 250 mm) with a mobile phase of methanol–water (90:10 v/v) in isocratic mode at a flow rate of 1 ml/min with 278 nm as the analytical wavelength and 20 μl injection volume (Farah et al., Citation2013; Khan et al., Citation2013). Similarly for PIP, analysis was carried out by RP-HPLC (Shimadzu UFLC model) on C-18 column Inertsil® (ODS-3 V, dimension 4.6 × 250 mm) with a mobile phase of acetonitrile–water (60:40 v/v) in isocratic mode at a flow rate of 1.5 ml/min with 343 nm as analytical wavelength and 20 μl injection volume (Chen et al., Citation2007). Calibration curves were prepared in the concentration range of 0.1–50 and 0.25–50 μg/ml for RPM and PIP, respectively. The entrapment efficiency was calculated by the formula given below:
Transmission electron microscopy
Transmission electron microscopy (TEM) analysis was performed to study the morphology of developed nanoparticles. Carbon-coated grids were pre-treated with nanoparticle suspension for 5–10 min. After washing, 2% uranyl acetate was used to stain the particles and grids were air dried. Particles were observed at various magnifications using Hitachi, H-7500 and TEM images were recorded.
Freeze thaw study
Freeze thaw study was carried out by subjecting nanoparticle dispersion to three freeze thaw cycles. The nanoparticle dispersion was exposed to three cycles comprised of freezing at −20 °C for half an hour in a deep freezer followed by thawing at 30 °C. The particle size and PDI before starting freeze thaw cycles and after completion of freeze thaw cycles were determined by zeta sizer. This serves as a parameter to analyse the physical stability of nanoparticles undergoing sudden temperature changes.
In vitro release from nanoparticles
In vitro release of RPM and PIP from nanoparticles was conducted using dialysis bag method, wherein pellets of nanoparticles obtained after centrifugation were resuspended in release media (0.5 ml) to form a nanoparticle suspension (RPM nanoparticles, PIP nanoparticles and co-encapsulated nanoparticles) equivalent to 0.5 mg of both drugs. The obtained suspension was taken inside a dialysis bag and placed in 9.5 ml of release media (Saline: Isopropanol [IPA], 90:10) to maintain sink conditions. It was kept in a constant shaker bath at 100 rpm at 37 °C (Khan et al., Citation2013). At regular time intervals, 8 ml of the release medium was sampled and replaced with 8 ml of the fresh release medium. Then samples were analyzed by HPLC. Same procedure was followed to check the release profiles of free drugs, i.e. RPM and PIP.
P-gp efflux study
The everted gut sac method was used to study the P-gp efflux of RPM in the presence or absence of PIP solution and in the form of nanoparticle formulation (Sha & Fang, Citation2004). Everted sacs of rat duodenum and ileum were prepared using the method described by Sasaki et al. (Citation1995). Female Sprague–Dawley (SD) rats weighing between 230 and 270 g were deprived of food for 1 day, and they were provided with only double distilled water. After obtaining the intestine (length 2 cm), it was everted inside out, and the sac was ligated. Different sacs were filled with 1 ml tyrode solution and placed in four different beakers containing 20 ml of buffer with the following formulations: RPM suspension, RPM suspension + PIP suspension, RPM nanoparticle and RPM + PIP nanoparticles (co-encapsulated) with proper aeration. The RPM concentration in the outer buffer was 100 µg/ml. The study was conducted for one and a half hours, and the internal content of the intestine at end point was analyzed for the RPM content by HPLC method (described above), to determine the extent of P-gp efflux inhibition.
Cell culture study
Human-derived breast cancer cell line (MDA-MB-231) from adenocarcinoma (cancer of breast epithelium tissue) was used in this study. MDA-MB-231 cells were seeded in 96-well plates at a density of 5000 viable cells per well and incubated 24 h to allow cell attachment (Noh et al., Citation2004). The cells were incubated with different formulations at 0.1, 1, 10, 20, 40, 100 μM/ml equivalent drug concentrations, and drug-free polymeric nanoparticle suspension as well with the same amount of polymer for 24 h. Afterwards, formulations were replaced with RPMI containing 3 -(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (250 μg/ml), and cells were then incubated for an additional 4 h. MTT was aspirated, and dimethylsulfoxide (DMSO) was added to dissolve the formazan crystals. Absorbance was measured at 570 nm using a microplate reader. Untreated cells were taken as control with 100% viability, and cells without addition of MTT were used as blanks to calibrate the spectrophotometer to zero absorbance. The drug concentration, at which inhibition of 50% cell growth (IC50) was observed in comparison with that of the control sample, was calculated by curve fitting of the cell viability data. Experiments were performed in triplicate, and results were expressed as mean ± standard deviation (SD).
Pharmacokinetic study
Pharmacokinetic experiments were performed on female SD rats. The rats were housed under controlled environmental conditions (temperature 23 ± 1 °C; humidity 55 ± 5%; 12 h light/dark cycles) with food and water ad libitum. Animal experiments were conducted according to institutional guidelines for the care and the use of laboratory animals, and the protocol was approved by the Institutional Animal Ethics Committee. Four groups of animals were used for the pharmacokinetic study. The groups were RPM suspension, RPM + PIP suspension, RPM nanoparticles, RPM + PIP nanoparticles (six animals in each group). Both drugs were administered at a dose level of 10 mg/kg once. Oral dosing was given for each group, and blood samples were collected at time intervals of 0.5, 2, 4, 8, 24, 48, 72, 120 and 168 h. These samples were subsequently extracted for drug content.
The extraction procedure of blood involves the following steps. In the tubes containing 0.2 ml of blood, 0.4 ml of sodium carbonate (0.1 M) and 5 µl of internal standard (cyclosporine, 1 mg/ml) were added and vortexed for 1 min. After vortexing, 10 ml of tert-butyl methyl ether was added. The tubes were again vortex-mixed for 1 min, and they were kept in shakers for 15 min. Thereafter, they were centrifuged at 4000 rpm for 3 min. Supernatants were air dried. The dried extracts were reconstituted in 300 µl of methanol, and samples were analyzed by RP-HPLC for RPM (Shimadzu UFLC model) on C-18 column Inertsustain® (ODS, dimension 4.6 × 150 mm) with a mobile phase of methanol–water (85:15% v/v) in isocratic mode at a flow rate of 1.2 ml/min with 278 nm as analytical wavelength and 20 μl injection volumes (Khan et al., Citation2013). RPM's retention time was observed at 4.63 min with these conditions and run time was extended to 10 min. Cyclosporin was used as an internal standard. Its measurement was carried out at 220 nm. A calibration curve was prepared in the concentration range of 0.1–1 μg/ml. Using the calibration curve the concentration of RPM in blood was determined. The blood concentration profile was plotted against time and pharmacokinetic parameters, such as Cmax, Tmax, AUC were calculated using the software Kinetica© (Thermo Scientific, Waltham, MS).
Results and discussion
Preparation and characterization of nanoparticles
Among the various methods used in the preparation of polymeric nanoparticles, the nanoprecipitation method was selected since no/less external energy input is essential for nanoparticle formation (i.e. required high shearing homogenization, milling or sonication). Thus, nanoprecipitation can be regarded as a mild and sensitive procedure with modest equipment requirements and low energy costs. In contrast to emulsion/solvent diffusion, no surfactants that might influence the surface characteristics or cause toxic effects (Schubert et al., Citation2011) are necessary. In this method, polymer and drug are dissolved in an organic phase, which is further added to an aqueous phase with/without surfactant to achieve finely precipitated nanoparticles. The mechanism of formation of nanoparticles by nanoprecipitation is explained by interfacial turbulence generated during the process of precipitation. Subsequently, a violent spreading is observed because of mutual miscibility between the solvents. Droplets of solvent, probably of nanometric size, are torn from the interface. These droplets are rapidly stabilized by the stabilizing agent, until diffusion of the solvent is complete, and polymer aggregation occurs (Quintanar-Guerrero et al., Citation1998). Chlorinated solvents have been extensively used with this method to dissolve the PLGA (i.e. methylene chloride, dichloromethane, chloroform), but their toxicity is of concern. A possible alternative solvent to chlorinate is acetone. The low toxicity and low boiling point (56 °C) are the main advantages of using acetone to dissolve the polymer. Since acetone is completely water miscible, it rapidly diffuses, leading to precipitation in nano form (Rao & Geckeler, Citation2011).
The amount and type of stabilizer used also have an effect on particle properties, wherein the surfactants avoid aggregation and stabilize nanoparticles (Chorny et al., Citation2002). This may be attributed to the surfactant properties, which have shown variability in this method. Surfactant types and concentrations were optimized. Similarly, polymer concentration was optimized based on balance between yield and particle size. Surfactants, such as PVA (2%), Tween 80 (2%), Tween 20 (2%) and Vitamin E TPGS (0.075%), were screened in the protocol. The surfactant concentration used in this experiment was based on the most reported mean values and all other parameters used in the experiment were fixed to a constant value (polymer concentration, organic solvent volume and homogenization time). Out of various screened surfactants, Tween 20 at a concentration of 2% gave the least particle size (104.9 ± 10.2 nm) with PDI 0.224; whereas those with Vitamin E TPGS, Tween 80 and PVA were resulted in particles of larger size (2683 ± 1120, 123.9 ± 13.9 and 197.7 ± 27.6 nm, respectively) and higher PDI (>0.3) in their normally used concentration ranges (). Once the surfactant was fixed as Tween 20, the surfactant concentration was varied at level of 1% and 2% keeping other parameters constant to study the effect of surfactant concentration on nanoparticles. As shown in , smaller particle size was obtained with 2% concentration range, which was fixed for further trials.
Figure 1. Results obtained from various studies involved in the preparation of polymeric nanoparticles. (a) Effect of different surfactants/concentration on particle size and PDI: Tween 20 at a concentration of 2% gave the least particle size. (b) Effect of polymer concentration: particle size was decreased with an increase in polymer concentration. (c) Size and entrapment efficiency of optimized, drug-loaded nanoparticles. (d) Nanoparticles exposed to three freeze thaw cycles at 30 and −20 °C and no significant change in size and PDI was observed. (e) Intensity-based size distribution report of co-encapsulated nanoparticles. (f) TEM image of prepared nanoparticles. Values are expressed as mean ± SD, n = 3.
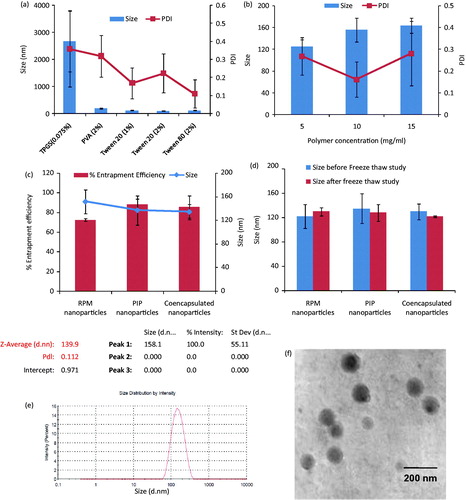
After fixing the surfactant type and concentration, effect of polymer amount on particle size was studied at three concentration levels of 5%, 10% and 15%. Particle size was found to be increased with an increase in polymer concentration from 5% to 15%, since same amount of surfactant cannot stabilize the increased amount of polymer, and resulting in larger particle size (Mainardes & Evangelista, Citation2005). The particle size for 5%, 10% and 15% were found to be 124.6 ± 12.5, 155.6 ± 21.8 and 163.4 ± 13.7 nm (PDI 0.268 ± 0.086, 0.161 ± 0.081 and 0.280 ± 0.148), respectively. Polymer concentration of 10% was chosen based on the size, PDI and yield which would be higher as compared to 5% polymer concentration. The optimized parameters were then utilized to prepare drug-loaded nanoparticles. The optimized parameters, size and PDI of nanoparticles are shown in .
Size and size distribution
Based on the fixed values of optimized parameters, individual and drug-loaded nanoparticles were prepared. Particle size was analyzed using dynamic light scattering technique. The size for RPM nanoparticles, PIP nanoparticles and co-encapsulated nanoparticles was determined to be 151.65 ± 19.73, 137.06 ± 24.50, 134.33 ± 12.78 nm and PDI 0.201 ± 0.085, 0.157 ± 0.129, 0.149 ± 0.011, respectively (). It is anticipated that with co-encapsulation of two drugs, the particle size has to be increased. In this case, however, the particle size was comparable to that of individual drug-loaded nanoparticles.
Entrapment efficiency
Entrapment efficiency was calculated by analyzing supernatant through HPLC. Nanoprecipitation method resulted in better entrapment of drugs in polymeric matrix. shows more than 70% entrapment of both the drugs in nanoparticles.
Transmission electron microscopy
Size of nanoparticles was further confirmed by TEM analysis. TEM images () revealed that nanoparticles are spherical in size with homogeneous particle size distribution.
Freeze thaw study
A freeze thaw study was conducted to check the stability of nanoparticles with drastic temperature changes (room temperature to −20 °C). Formulations were found to be stable for three freeze thaw cycles in terms of size and PDI. Particle sizes of less than 150 nm were observed before and after freeze thaw for all formulations. The particle size and PDI before and after the cycles did not show significant variation, but some degree of precipitation was observed with each formulation. This may be due to interaction of particles with each other during the step of freezing resulting into clumping or aggregation of particles, but the degree of such aggregation was less. The particle size and PDI of nanoparticles before and after the freeze thaw cycle are given in .
In vitro drug release from nanoparticles
Drug release from a PLGA matrix occurs through diffusion-cum-degradation mediated process. During the early phases, release occurs mainly through diffusion in the polymer matrix, while during later phases release is mediated through both diffusion of the drug and degradation of the polymer matrix, itself. In vitro release of RPM and PIP was determined from individual nanoparticles and co-encapsulated nanoparticles containing both the drugs. The rate of release from PLGA nanoparticles is slow, which can be attributed to the rigid chain structure of PLGA and its rate of degradation (Panyam et al., Citation2003; Hariharan et al., Citation2006). Generally, phosphate buffer saline (PBS) with pH 7.4 is employed as a release medium to study in vitro release of nanoparticles. RPM is reported to be unstable in PBS due to alkaline hydrolysis of lactam ring leading to ring opening (Nelson et al., Citation1999). Although the solubility of RPM is very low in a buffer, it is sufficient to undergo hydrolysis. Reported data suggest the stability of RPM in 9:1 (v/v) normal saline and isopropanol sufficient enough to carry out a release study (Naseerali et al., Citation2010).
Nearly 100% release was observed in case of both RPM solution and PIP solution (free drug form). The release of RPM was found to be slow as compared to PIP from the nanoparticles. Almost complete release of PIP was observed in 14 days, whereas the release of RPM was slowed down in the presence of PIP (only 76% release of RPM from co-encapsulated nanoparticles in 49 days and 90% release from RPM nanoparticles in 49 days), but release of PIP was not effected in co-encapsulated nanoparticles. This release pattern shows some degree of initial burst release followed by sustaining the release profile. The release profiles of RPM, PIP from individual and co-encapsulated nanoparticles are shown in , respectively and release from drug solutions is shown in . Since the release is in a biphasic pattern, it is divided into two parts, where 0–3 h is considered as burst release phase; 3 h and onwards the sustained release pattern is evaluated. The release pattern was studied for zero order, first order and Higuchi kinetics. The Peppas model was applied to study the release mechanism. Kinetic profiling indicates zero order release with non-Fickian transport in the initial phase and subsequently Higuchi kinetics with Fickian transport during the remaining phase ().
Figure 2. In vitro release profiles of RPM and PIP from (a) individually drug-loaded nanoparticles and (b) co-encapsulated drug nanoparticles and (c) free drug solutions. Study was carried out in dialysis membrane at 37 °C, and normal saline: IPA (90:10, %v/v) was used as release medium.
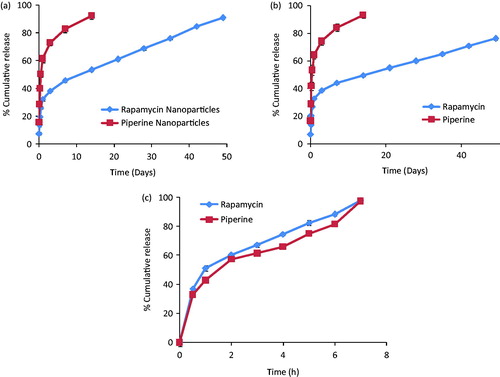
Table 1. Drug release profile and kinetics from individual and co-encapsulated nanoparticles.
P-gp efflux study
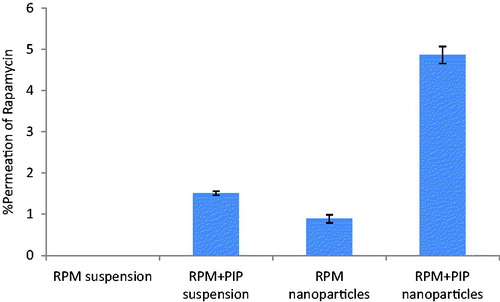
The everted gut sac technique is a simple and effective method to study absorptive transport and the action of intestinal P-gp on intestinal drug absorption in vitro (Cornaire et al., Citation2004). As most of the carriers including P-gp receptors are present on mucosal side, gut sacs are everted for better results (Elsheikh et al., Citation2014). Drugs absorbed from the mucosal to the serosal side were calculated. Detectable amounts of RPM were not found inside the gut sac within 90 min of the study period, when it was in suspension form. P-gp efflux can be one of the possible hindrances in the transport of RPM from mucosal to serosal side. Better permeation of RPM (∼1%) in the form of nanoparticles was observed (). The potential of nanocarriers in modulating P-gp can be attributed to this. Tween 20 surfactant used in the preparation of RPM-loaded nanoparticles also possesses a P-gp inhibition property (Nieto Montesinos et al., Citation2012). Hence, absorption of RPM was improved. Permeation of RPM in the presence of PIP solution (∼5%) was much more than that of nanoparticles. Inhibition of P-gp efflux by PIP resulted in increased transport of RPM across the gut membrane (Bhardwaj et al., Citation2002). When both RPM and PIP were tested in the form of nanoparticles, a 5-fold increase in RPM absorption was observed. Presence of PIP (P-gp inhibitor) and encapsulation of RPM in the form of nanoparticles prevent the efflux mechanism and help in reaching the drug inside the gut sac.
Figure 3. Percent permeation of RPM over intestinal barrier using everted gut sac method. Gut sacs were maintained with proper aeration for a period of 90 min. Effect of PIP as a Pgp modulator and potential of nanoparticulate delivery system on intestinal transport of RPM were studied.
Cell culture study
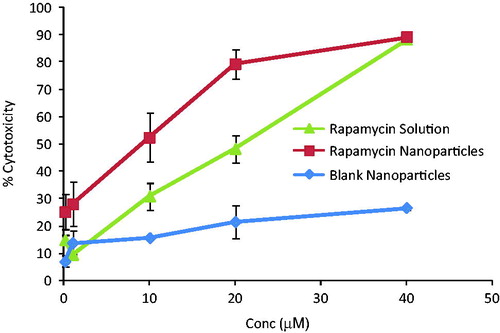
Cell viability assay depends on the ability of living cells to convert MTT into purple colored formazan crystals, which can be detected by measuring absorbance at 570 nm using a spectrophotometer. MDA-MB-231 breast cancer cell lines were used in this study. These cells are derived from human adenocarcinoma (cancer of breast epithelium tissue). Higher concentrations of RPM were employed, since these cell lines are more resistant to drug treatment (Chen et al., Citation2003). RPM solution and nanoparticles were added to cells at serial dilutions ranging from 0.1 to 40 µM of equivalent RPM. Concentration-dependent growth suppression was observed for both solution and nanoparticles of RPM. Nanoparticles were more effective in killing cancer cells in comparison with solution. The effect of blank nanoparticles on cell lines was also studied to confirm that nanoparticles as such do not cause cytotoxicity without drug. An IC50 of 20.35 µM was observed for RPM solution. For RPM nanoparticles it was 11.39 µM at 24 h (), indicating a higher efficacy of drug-loaded nanoparticles over solution. Sensitivity of MDA-MB-231 cell lines to RPM treatment was reported between 2 and 20 µM (Yellen et al., Citation2011). Cells were also incubated with PIP, 24 h prior to drug treatment to check the efficacy of RPM in the presence of chemosensitizer. No significant difference in cell viability was observed with PIP co-treatment.
Figure 4. Percent cytotoxicity of different concentrations of rapamycin solution, rapamycin-loaded nanoparticles and blank nanoparticles. Cells were incubated with these formulations for 24 h. Drug-loaded nanoparticles were more efficacious in killing breast cancer cells.
Pharmacokinetic study
This study is mainly aimed to check the effect of PIP, which acts as an absorption enhancer/chemosensitizer on oral absorption of RPM. RPM exhibits a tendency to partition into the red blood cells (RBCs) (38 times more concentrated into the RBCs) (Yatscoff et al., Citation1995). Thus, it becomes necessary to perform whole blood analysis instead of plasma analysis; since the sensitivity of the method would be lost due to lower concentration of RPM in plasma. PIP is reported to be a well-known P-gp inhibitor that this forms the basis of kinetics in combination of two drugs. Like most of the anticancer drugs, RPM is a P-gp substrate. Hence, most of the tumor cells that are rich in P-gp efflux pumps pump out the RPM. Use of PIP as absorption enhancer is expected to inhibit P-gp efflux, finally leading to the accumulation of RPM inside tumor cells (Shaikh et al., Citation2009). When the same phenomenon is applied, PIP is expected to inhibit P-gp receptors present on the intestinal barrier, finally allowing RPM to permeate across the intestinal barrier.
Blood levels of RPM after oral administration of four different formulations [i.e. (a) RPM suspension, (b) RPM suspension + PIP suspension, (c) RPM nanoparticles, (d) RPM nanoparticles + PIP nanoparticles] were analyzed and plotted against time. The mean RPM concentrations in the blood after oral administration of different formulations are given in and . For the formulations which were administered in the form of suspension, sharp Cmax was observed at Tmax 24 h. RPM suspension in the presence of co-administered PIP suspension resulted in Cmax of 15.34 μg/ml, which is the highest mean blood concentration of RPM among all the four formulations. An almost 3-fold increase in the highest mean blood concentration of RPM was observed in the presence of PIP suspension. Due to the sustained release of drugs from nanoparticles, Cmax for two formulations was observed at 72 h of Tmax. Interestingly, the effect of PIP nanoparticles in improving Cmax of RPM was not observed here. AUC values of all formulations were calculated. There is a 1.7-fold increase in bioavailability of RPM in the presence of a PIP suspension. More than a 3.5-fold increase in bioavailability was observed in RPM nanoparticles as compared to RPM suspension. The PIP effect on absorption was observed in nanoparticles with an approximate 4.8-fold increase in bioavailability compared to RPM suspension. The pharmacokinetic parameters of different formulations are given in , which shows considerable differences in bioavailability.
Figure 5. Mean blood concentration–time profiles of rapamycin in female SD rats following oral administration of (a) RPM suspension, (b) RPM-loaded nanoparticles, (c) RPM suspension and PIP suspension and (d) RPM-loaded nanoparticles and PIP-loaded nanoparticles at a single dose of 10 mg/kg for both the drugs (mean ± SD, n = 6).
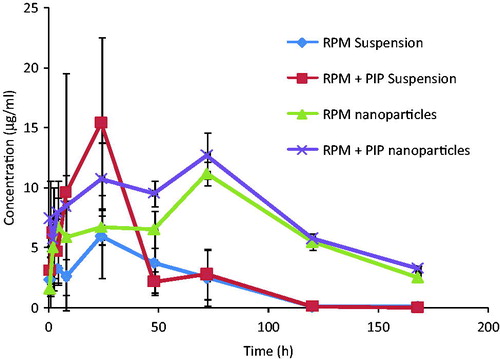
Table 2. Pharmacokinetic parameters of RPM in rats following oral administration of different formulations at a dose of 10 mg/kg for both the drugs (mean ± SD, n = 6).
Conclusions
PLGA polymeric nanoparticles of the anticancer drug RPM and chemosensitizer PIP (also possessing anticancer activity) were prepared and optimized. Nanoparticles were characterized for size, PDI and entrapment efficiency. Results obtained with co-encapsulated nanoparticles were almost comparable with that of individually drug loaded nanoparticles in every experiment. Sustained release of RPM until 49 days and PIP until 14 days was observed, suggesting low-dosing frequency. The potential of nanoparticles in the oral delivery of anticancer drugs was proven, as RPM absorption was more through gut sac in the form of nanoparticles. The presence of PIP also made an impact in improving the absorption of RPM, contributing to its P-gp inhibitory activity. Cytotoxicity studies on MDA-MB-231 cell lines suggest improved efficacy of RPM loaded nanoparticles in killing breast cancer cells. An in vivo pharmacokinetic study shows improvement in bioavailability of RPM in presence of PIP (4.8-folds), and also that the nanoparticles exhibit sustained release, thus having potential for long-term therapeutic action with less dosing frequency. Thus, the study suggests that the combination of RPM and PIP would result in reduction of dosing and improved bioavailability compared to single drug administration. Moreover, the two different pathways for the action of two anticancer drugs would help achieve a synergistic effect. An additional advantage of PIP is that P-gp efflux inhibitor and permeation enhancer increases the penetration and local concentration of drugs at the tumor site. Therefore, it is speculated that the combination of RPM and PIP nanoparticles might be more effective in treatment of breast cancer than single drugs in solution or nanoparticles in formulation.
Declaration of interest
The authors state no conflict of interest in preparation of this manuscript.
The authors acknowledge Ministry of Chemicals and Fertilizers, Government of India, and the Project Director, NIPER Hyderabad for providing a fellowship and funding for the project. Director CRIUM, Hyderabad duly acknowledged for kind support. Indo Vedic Nutrients Pvt. Ltd. is to be thanked for providing a gift sample of PIP.
References
- Abu-Khalaf M. (2014). A study of the mTOR inhibitor rapamycin (Rapamune, Sirolimus) in combination with abraxane in advanced solid cancers [online]. National Institute of Health, US. Available from: http://clinicaltrials.gov/show/NCT00337376 [last accessed 11 Oct 2014]
- Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. (2003). P-glycoprotein: from genomics to mechanism. Oncogene 22:7468–85
- Bezerra DP, Castro FO, Alves A, et al. (2006). In vivo growth-inhibition of Sarcoma 180 by piplartine and piperine, two alkaloid amides from Piper. Braz J Med Biol Res 39:801–7
- Bhardwaj RK, Glaeser H, Becquemont L, et al. (2002). Piperine, a major constituent of black pepper, inhibits human P-glycoprotein and CYP3A4. J Pharmacol Exp Ther 302:645–50
- Borst P, Zelcer N, Van de Wetering K, Poolman B. (2006). On the putative co-transport of drugs by multidrug resistance proteins. FEBS Lett 580:1085–93
- Chang J, Jallouli Y, Kroubi M, et al. (2009). Characterization of endocytosis of transferrin-coated PLGA nanoparticles by the blood brain barrier. Int J Pharm 379:285–92
- Chen Y, Brill GM, Benz NJ, et al. (2007). Normal phase and reverse phase HPLC-UV-MS analysis of process impurities for rapamycin analog ABT-578: application to active pharmaceutical ingredient process development. J Chromatogr B 858:106–17
- Chen Y, Zheng Y, Foster DA. (2003). Phospholipase D confers rapamycin resistance in human breast cancer cells. Oncogene 22:3937–42
- Chorny M, Fishbein I, Danenberg HD, Golomb G. (2002). Lipophilic drug loaded nanospheres prepared by nanoprecipitation: effect of formulation variables on size, drug recovery and release kinetics. J Control Release 83:389–400
- Cornaire G, Woodley J, Hermann P, et al. (2004). Impact of excipients on the absorption of P-glycoprotein substrates in vitro and in vivo. Int J Pharm 278:119–31
- Das S, Haddadi A, Veniamin S, Samuel J. (2008). Delivery of rapamycin-loaded nanoparticle down regulates ICAM-1 expression and maintains an immunosuppressive profile in human CD34+ progenitor-derived dendritic cells. J Biomed Mater Res A 85:983–92
- Elsheikh MA, Elnaggar YSR, Abdallah OY. (2014). Rationale employment of cell culture versus conventional techniques in pharmaceutical appraisal of nanocarriers. J Control Release 194:92–102
- Farah S, Khan W, Domb AJ. (2013). Crystalline coating of rapamycin onto a stent: process development and characterization. Int J Pharm 445:20–8
- Ganta S, Amiji M. (2009). Coadministration of paclitaxel and curcumin in nanoemulsion formulations to overcome multidrug resistance in tumor cells. Mol Pharm 6:928–39
- Haddadi A, Elamanchili P, Lavasanifar A, et al. (2008). Delivery of rapamycin by PLGA nanoparticles enhances its suppressive activity on dendritic cells. J Biomed Mater Res A 84:885–98
- Hariharan S, Elamanchili P, Lavasanifar A, et al. (2006). Design of estradiol loaded PLGA nanoparticulate formulations: a potential oral delivery system for hormone therapy. Pharma Res 23:184–95
- Ho EA, Soo PL, Allen C, Piquette-Miller M. (2007). Impact of intraperitoneal, sustained delivery of paclitaxel on the expression of P-glycoprotein in ovarian tumors. J Control Release 117:20–7
- Hoffman A, Domb AJ, Elgart A, Cherniakov I. (2013). Formulation and method for increasing oral bioavailability of drugs. PCT/IL2013/050047
- Khan W, Farah S, Nyska A, Domb AJ. (2013). Carrier free rapamycin loaded drug eluting stent: in vitro and in vivo evaluation. J Control Release 168:70–6
- Mainardes RM, Evangelista RC. (2005). PLGA nanoparticles containing praziquantel: effect of formulation variables on size distribution. Int J Pharm 290:137–44
- Naseerali CP, Hari PR, Sreenivasan K. (2010). The release kinetics of drug eluting stents containing sirolimus as coated drug: role of release media. J Chromatogr B 878:709–12
- Nelson FC, Stachel SJ, Eng CP, Sehgal SN. (1999). Manipulation of the C (22)–C (27) region of rapamycin: stability issues and biological implications. Bioorg Med Chem Lett 9:295–300
- Nieto Montesinos R, Beduneau A, Pellequer Y, Lamprecht A. (2012). Delivery of P-glycoprotein substrates using chemosensitizers and nanotechnology for selective and efficient therapeutic outcomes. J Control Release 161:50–61
- Noh W-C, Mondesire WH, Peng J, et al. (2004). Determinants of rapamycin sensitivity in breast cancer cells. Clin Cancer Res 10:1013–23
- Panyam J, Dali MM, Sahoo SK, et al. (2003). Polymer degradation and in vitro release of a model protein from poly(d,l-lactide-co-glycolide) nano-and microparticles. J Control Release 92:173–87
- Patil Y, Sadhukha T, Ma L, Panyam J. (2009). Nanoparticle-mediated simultaneous and targeted delivery of paclitaxel and tariquidar overcomes tumor drug resistance. J Control Release 136:21–9
- Quintanar-Guerrero D, Allemann E, Fessi H, Doelker E. (1998). Preparation techniques and mechanisms of formation of biodegradable nanoparticles from preformed polymers. Drug Dev Ind Pharm 24:1113–28
- Rao JP, Geckeler KE. (2011). Polymer nanoparticles: preparation techniques and size-control parameters. Prog Polym Sci 36:887–913
- Sahoo SK, Labhasetwar V. (2005). Enhanced antiproliferative activity of transferrin-conjugated paclitaxel-loaded nanoparticles is mediated via sustained intracellular drug retention. Mol Pharma 2:373–83
- Sasaki I, Tanaka K, Fujita T, et al. (1995). Intestinal absorption of azetirelin, a new thyrotropin-releasing hormone (TRH) analogue. II. In situ and in vitro absorption characteristics of azetirelin from the rat intestine. Bio Pharma Bull 18:976–9
- Schubert S, Delaney Jr JT, Schubert US. (2011). Nanoprecipitation and nanoformulation of polymers: from history to powerful possibilities beyond poly (lactic acid). Soft Matter 7:1581–8
- Seto B. (2012). Rapamycin and mTOR: a serendipitous discovery and implications for breast cancer. Clin Transl Med 1:1–7
- Sha X, Fang X. (2004). Transport characteristics of 9-nitrocamptothecin in the human intestinal cell line Caco-2 and everted gut sacs. Int J Pharm 272:161–71
- Shaikh J, Ankola DD, Beniwal V, et al. (2009). Nanoparticle encapsulation improves oral bioavailability of curcumin by at least 9-fold when compared to curcumin administered with piperine as absorption enhancer. Eur J Pharm Sci 37:223–30
- Shoba G, Joy D, Joseph T, et al. (1998). Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med 64:353–6
- Simamora P, Alvarez JM, Yalkowsky SH. (2001). Solubilization of rapamycin. Int J Pharm 213:25–9
- Song XR, Cai Z, Zheng Y, et al. (2009). Reversion of multidrug resistance by co-encapsulation of vincristine and verapamil in PLGA nanoparticles. Eur J Pharma Sci 37:300–5
- Vijayakumar RS, Surya D, Nalini N. (2004). Antioxidant efficacy of black pepper (Piper nigrum L.) and piperine in rats with high fat diet induced oxidative stress. Redox Rep 9:105–10
- Yatscoff RW, Boeckx R, Holt DW, et al. (1995). Consensus guidelines for therapeutic drug monitoring of rapamycin: report of the consensus panel. Ther Drug Monit 17:676–80
- Yellen P, Saqcena M, Salloum D, et al. (2011). High-dose rapamycin induces apoptosis in human cancer cells by dissociating mTOR complex 1 and suppressing phosphorylation of 4E-BP1. Cell Cycle 10:3948–56