
Abstract
Biodegradable polymer nanoparticle drug carriers are an attractive strategy for oral delivery of peptide and protein drugs. However, their ability to cross the intestinal epithelium membrane is largely limited. Therefore, in the present study, cell-penetrating peptides (R8, Tat, penetratin) and a secretion peptide (Sec) with N-terminal stearylation were introduced to modify nanoparticles (NPs) on the surface to improve oral bioavailability of peptide and protein drugs. In vitro studies conducted in Caco-2 cells showed the value of the apparent permeability coefficient (Papp) of the nanoparticles co-modified with Sec and penetratin (Sec-Pen-NPs) was about two-times greater than that of the nanoparticles modified with only penetratin (Pen-NPs), while the increase of transcellular transport of nanoparticles modified together with Sec and R8 (Sec-R8-NPs), or Sec and Tat (Sec-Tat-NPs), was not significant compared with nanoparticles modified with only R8 (R8-NPs) or Tat (Tat-NPs). Using insulin as the model drug, in vivo studies performed on rats indicated that compared to Pen-NPs, the relative bioavailability of insulin for Sec-Pen-NPs was 1.71-times increased after ileal segments administration, and stronger hypoglycemic effects was also observed. Therefore, the nanoparticles co-modified with penetratin and Sec could act as attractive carriers for oral delivery of insulin.
Introduction
In recent years, oral delivery of peptide and protein drugs has remained to be a great challenge owing to their poor stability in the gastrointestinal tract and low permeability through the intestinal epithelium membrane caused by their high molecular weights and hydrophilicity inherently (Goldberg & Gomez-Orellana, Citation2003; Bernkop-Schnurch & Schmitz, Citation2007). To improve bioavailability of these drugs by oral administration, various promising strategies have been utilized to circumvent these obstacles (Renukuntla et al., Citation2013). These strategies include using enzyme inhibitors (Bernkop-Schnurch, Citation1998; Su et al., Citation2012) and drug delivery systems (Mahato et al., Citation2003; Fan et al., Citation2014) to protect these drugs from enzymatic degradation, as well as chemical absorption enhancers (Gupta et al., Citation2013) directing at improving intestinal epithelium membrane permeability. However, many of these approaches have been achieved limited success due to their drawbacks inherently. For example, co-administration of enzyme inhibitors for a long duration may induce unwanted side effects (Sood & Panchagnula, Citation2001).
With the development of nanotechnology, biodegradable polymer nanoparticle drug carriers have been considered as highly promising tools to improve oral bioavailability of peptide and protein drugs, as nanoparticles (NPs) possess the ability not only to improve the stability of encapsulated drugs but also to increase their retention time in the gastrointestinal tract (des Rieux et al., Citation2006; Gamboa & Leong, Citation2013). As the most commonly utilized polymer, poly(d,l-lactic-coglycolic-acid) (PLGA) has manifested the advantage of favorable biodegradability and biocompatibility and was approved by the FDA for pharmaceutical excipients (Panyam & Labhasetwar, Citation2003; Kumari et al., Citation2010). However, the ability of nanoparticles to cross the intestinal epithelium membrane is limited (Koren & Torchilin, Citation2012). Thus, in the present study, cell-penetrating peptides (CPPs) and Sec (an Engrailed secretion peptide) were harnessed to modify PLGA nanoparticles in order to overcome this obstacle.
Cell-penetrating peptides are typically described as cationic and/or amphipathic peptides containing 5–30 amino acids, which are mainly derived from natural proteins (such as penetratin and Tat) or artificial sequences like octaarginine and transportan (Patel et al., Citation2007; Stewart et al., Citation2008). Recently, they have received considerable interest as attractive delivery vehicles owing to their ability to cross the cellular membrane and mediate the uptake of cargoes into cells (Farkhani et al., Citation2014). Numerous studies have been reported concerning the application of CPPs to deliver a large variety of cargoes intracellularly, such as nanocarriers (Sawant et al., Citation2006; Sethuraman et al., Citation2008; Torchilin, Citation2008), quantum dots (Ruan et al., Citation2007), peptides and proteins (Liu et al., Citation2013a). Additionally, CPPs have been utilized to traverse intact cellular barriers, such as blood brain barrier (BBB) and intestinal epithelium barriers (Patel et al., Citation2007; Foged & Nielsen, Citation2008). Tat-modified PLGA nanoparticles showed great potential for brain delivery of insulin (Yan et al., Citation2012). The permeability of insulin and Tat peptide conjugate across Caco-2 cells monolayer was 6- to 8-fold increased in comparison with insulin alone (Liang & Yang, Citation2005).
In general, cellular internalization and secretion are both required for the transcellular transport. It was reported that the SecPen polypeptide combining Sec and penetratin (Pen) was able to efficiently cross the MDCK epithelium (Dupont et al., Citation2007). The explanation for the high transport efficiency was that Pen facilitated the step of internalization, whereas, Sec contributed to the step of secretion. The Sec peptide (QSLAQELGLNERQIKI), a 16 amino acid sequence derived from the second and third helix of the Engrailed homeodomain, was essential for Engrailed secretion. Based on such a background, we speculated that transcellular transport of peptide and protein drugs in intestinal enterocytes might be improved by using the Sec peptide together with CPPs, since Sec might promote the exocytosis of the cargoes after internalization.
Therefore, in this study, we designed a novel biodegradable polymer nanoparticle drug carrier co-modified with two kinds of cellular transport peptides including CPPs (R8, Tat, penetratin) and Sec for the oral delivery of peptide and protein drugs. Insulin was selected as the representative model drug. To our knowledge, there are no reports about modification of CPPs together with Sec on the surface of nanoparticles for the delivery of peptide and protein drugs. The transcellular transport, cellular uptake and cytotoxicity studies were conducted in human colon carcinoma cells (Caco-2 cells). To assess the effects of the two peptides modification on ileal insulin absorption, the pharmacokinetic and pharmacodynamic profiles were further investigated in rats.
Materials and methods
Materials and animals
N-terminally stearylated peptides (), including Stearyl-R8 (STR-R8), Stearyl-Tat (STR-Tat), Stearyl-penetratin (STR-Pen), Stearyl-Sec (STR-Sec), were synthesized by GL Biochem Co., Ltd. (Shanghai, China). PLGA (LA:GA 50:50, MW 8000, COOH terminated) was provided by Shandong Institute of Medical Instruments (Shandong, China). Recombinant pig insulin (28.7 IU/mg) was obtained from Xuzhou Wanbang Biochemical Pharmaceutical Co., Ltd. (Jiangsu, China). Soybean Phospholipid (PC≥90%) was supplied by Shanghai Taiwei Pharmaceutical Co., Ltd. (Shanghai, China). Sugar Ester S-1670 (SE) was kindly supplied by Mitsubishi-Kagaku Foods Co. (Tokyo, Japan). Polyvinyl alcohol (Kuraray PVA-124, MW 105 000) was obtained from Guangzhou Dingfengsheng Chemical Technology Co., Ltd. (Guangdong, China). Coumarin-6 was purchased from Sigma (St. Louis, MO). Cell Counting Kit-8 (CCK-8), BCA Protein Quantification Kit and RIPA lysis buffer were purchased from Vazyme Biotech Co., Ltd. (Nanjing, China). Rat INS (Insulin) ELISA Kit was purchased from Elabscience Biotechnology Co., Ltd. (Wuhan, China). A glucose meter (ACCU-CHEK® Performa) was purchased from Roche Pharma Ltd. (Shanghai, China). All other chemical reagents were of analytical grade or HPLC grade.
Table 1. Sequences of N-terminally stearylated CPPs and Sec used in this study.
Cell culture medium RPMI-1640, 0.25% trypsin-0.02% EDTA, phosphate buffer solution (PBS), Hank’s balanced salt solution (HBSS) were all purchased from Solarbio Technology Co., Ltd. (Beiing, China). Fetal bovin serum (FBS) was obtained from TransGen Biotech Co., Ltd. (Beiing, China).
Male Sprague–Dawley rats weighing 180 ± 20 g, 11 weeks old, were provided by the Experimental Animal Center of Nanchang University. The rats were housed under standard conditions at a room temperature of 22–24 °C and humidity of 40–60% with free access to food and water supply.
Preparation of nanoparticles
Preparation of insulin–phospholipid complex
Insulin–phospholipid (Ins-SPC) complex was prepared according to the previous report (Cui et al., Citation2006). In brief, 40 mg of insulin and 600 mg of phospholipid were dissolved in 15 mL dimethyl sulfoxide (DMSO) containing 5% glacial acetic acid. The mixture was stirred in a water bath at 30 °C to obtain a clear solution, and then freeze-dried for 24 h to remove the solvent.
Determination of residual DMSO
Residual DMSO was measured using a gas chromatograph (GC) system (Agilent 6890N, Santa Clara, CA) equipped with flame ionization detector (FID) and a DB-624 (0.53 mm × 30 m × 3.00 μm) column. The inlet split ratio was 5:1 and the carrier gas was nitrogen at a velocity of 2.0 mL/min. Ins-SPC complex was dissolved in anhydrous ethanol. Samples (1 μL) were injected into the GC system with the column temperature of 150 °C.
Preparation of nanoparticles
Insulin-loaded nanoparticles modified with N-terminally stearylated CPPs (STR-CPPs) were prepared by using the emulsion solvent evaporation method (Cui et al., Citation2006). Briefly, 30 mg of PLGA and 20 mg of Ins-SPC complex were dissolved in 700 μL dichloromethane and 300 μL ethyl acetate as the oil phase. The water phase was formulated by adding 5 mg STR-CPPs into 5 mL of 0.3% SE aqueous solution. Then the oil phase was poured into the water phase under sonication at 550 W for 2 min in an ice-water bath to form the oil-in-water emulsion. Subsequently, the organic solvent that existed in emulsion was removed under magnetic stirring for at least 4 h. The resulting nanoparticle suspensions were centrifuged at 20 000 rpm and 4 °C for 20 min by an ultracentrifuge (Optima L-100K, Beckman Coulter, Brea, CA), repeatedly washed with distilled water for three times and finally resuspended in distilled water.
Insulin-loaded nanoparticles co-modified with STR-Sec and STR-CPPs were prepared by the same procedure, except that 5 mg STR-CPPs and 5 mg STR-Sec were added into the 0.3% SE aqueous solution.
Coumarin-6-loaded nanoparticles were used for the cell association studies owing to intrinsic fluorescence of coumarin-6. The nanoparticles were formulated by substituting coumarin-6 for Ins-SPC complex. The free coumarin-6 was removed by centrifugation at 20 000 rpm and 4 °C for 20 min. The coumarin-6 concentration in all test nanoparticle suspensions was 1 µg/mL. Unmodified nanoparticles were prepared by the same approach, while the water phase was 3% PVA aqueous solution.
Characterization of nanoparticles
The morphology of nanoparticles was observed by using transmission electron microscopy (JEM-2100, JEOL, Tokyo, Japan). Briefly, nanoparticle samples after dilution were dropping onto copper grids, and then negatively stained by 2% phosphotungstic acid before measurement. The particle size and zeta potential of nanoparticles were further detected using Malvern Mastersizer (PSA NANO2590, Malvern Instruments, Malvern, UK).
The encapsulation efficiency (EE) and drug loading (DL) of coumarin-6-loaded nanoparticles were measured by using fluorescence spectrometer (PerkinElmer LS55, Waltham, MA) at 450 nm excitation wavelength and 502 nm emission wavelength. Coumarin-6 amount in the supernatant was quantified after centrifugation.
For insulin-loaded nanoparticles, the concentration of free insulin in the supernatant was measured using HPLC (Agilent 1260 Infinity, Santa Clara, CA) method. The system contained a 1260-Quaternary pump, a Hypersil ODS2 column (4.6 mm × 250 mm, 5 µm), and a 1260-VWD UV/Vis detector. The mobile phase was composed of 0.2 M sulfate buffer solution (pH 2.3) and acetonitrile with the ratio of 74:26. The flow rate was kept at 1.0 mL/min and the detection wavelength was 214 nm. The EE and DL of nanoparticles were calculated as the following equations:
(1)
(2)
In vitro leakage study of coumarin-6
The in vitro release profiles of coumarin-6 from nanoparticles were investigated to determine whether coumarin-6 would serve as an effective fluorescent probe. Firstly, 2 mL of nanoparticle suspensions was placed in a dialysis bag (MWCO = 14 kDa). The dialysis bag was firmly sealed and transferred to an iodine flask containing 48 mL of phosphate buffer solution (PBS). Then the iodine flask was located in a water-bath shaker at constant shaking of 100 rpm at 37 °C. After an aliquot (1 mL) of the release medium was withdrawn at defined intervals, the equivalent volume of fresh medium was replenished immediately. Phosphate buffer solution with pH 7.4 and pH 4.0 values were tested as the release medium. The released coumarin-6 concentration was determined as described above. The cumulative release percentage of coumarin-6 was calculated.
In vitro release study of insulin
To investigate the in vitro release profiles of insulin from nanoparticles, insulin-loaded nanoparticles were resuspended in simulated intestinal medium (PBS, pH 6.8) without trypsin at 37 °C under constant shaking of 60 rpm. After an aliquot (1 mL) of the release medium was withdrawn at defined intervals, the equivalent volume of fresh medium was replenished immediately. Samples were added into ultrafiltration centrifugal tubes (MWCO = 100 kDa, Millipore, Billerica, MA) and centrifuged at 3500 rpm for 15 min. The filtrate was collected to determine the released insulin concentration by the HPLC method. The cumulative release percentage of insulin was calculated.
Enzymatic stability study
To assess the stability of insulin in the nanoparticles in intestinal fluids, insulin-loaded nanoparticles were resuspended in simulated intestinal medium (PBS, pH 6.8) with trypsin (0.4%, w/v) at 37 °C under constant shaking of 60 rpm. Free insulin solution was used as control. Samples were withdrawn at defined intervals and the enzymatic degradation reaction was terminated by the addition of 0.1 M HCl. The precipitation was obtained by centrifugation at 20 000 rpm and 4 °C for 20 min. 50 µL acetonitrile was added to destroy nanoparticles and 150 µL 0.1 M HCl was added to dissolve insulin. The concentration of insulin was determined by the HPLC method.
Cell culture
Caco-2 cells were cultured in 25 cm2 culture dishes in RPMI-1640 medium consisting of 10% of fetal bovine serum, 100 U/mL of penicillin, 100 mg/mL of streptomycin at 37 °C with 5% CO2 supply. The culture medium was exchanged every day by a fresh medium. After reaching 80–90% confluence, the cells were then subcultured with 0.25% trypsin–0.02% EDTA. Cells with passage numbers of 28–35 were used.
Transcellular transport study
For the transcellular transport study, Caco-2 cells were seeded onto polycarbonate membranes in 12-well transwell plates (3 μm pore size; Corning Costar, Corning, NY) at a density of 1 × 105 cells/cm2. The cells were cultivated for 21–28 days to form the monolayers. In the first 7 days, the culture medium was changed every other day, while for the next two weeks, the medium was changed every day. The transepithelial electrical resistance (TEER), an indicator for structural and functional integrity of the monolayers, was measured using the Millicell ERS-2 system (Millipore, Billerica, MA). The monolayers with TEER values above 500 Ω cm2 were selected for the transcellular transport study. Before the transport experiments, the monolayers were washed twice with Hank’s balanced salt solution (HBSS, pH 7.4) and equilibrated with transport buffer (HBSS, pH 7.4) for 30 min at 37 °C. And then 0.5 mL of coumaine-6-loaded nanoparticle suspensions, dispersed in HBSS, was added to the apical side. The basal chamber of the monolayers was immersed in 1.5 mL of HBSS. Following co-incubation of nanoparticles with the monolayers at defined intervals (0.5, 1, 2 and 4 h), an aliquot (0.2 mL) of the sample was withdrawn from the basal side to determine the total amount of permeated nanoparticles. Immediately, the equivalent volume of fresh HBSS was replenished. Collected samples were dispersed in acetone to destroy nanoparticles and then coumarin-6 amount in the samples was determined as described above.
The apparent permeability coefficient (Papp, cm/s) of nanoparticles was calculated using the following equation:
(3)
where dQ/dt is the linear appearance rate of nanoparticles transported across the monolayers (ng/s), A is the membrane area of the insert (cm2), and C0 is the initial concentration of encapsulated coumarin-6 added to the apical side (ng/mL).
Cellular uptake study
Caco-2 cells were seeded in 12-well plates at a density of 1 × 105 cells/cm2. The cells were cultivated in the culture medium as described above and incubated in a culture incubator at 37 °C, 5% CO2. Prior to the cellular uptake study, the cells were washed twice with HBSS and equilibrated with HBSS for 30 min. Subsequently, the cells were treated with coumaine-6-loaded nanoparticle suspensions for defined intervals (1, 2, 3 and 4 h). After co-incubation with nanoparticles, the cells were washed at least thrice with cold HBSS to terminate the cellular uptake and eliminate the surface-bound nanoparticles. In terms of the qualitative analysis, the cells were observed by fluorescence microscopy (Olympus, Tokyo, Japan). To quantify the uptake of nanoparticles, the cells were treated as mentioned above, then added with 0.25% trypsin-0.02% EDTA, and finally collected by centrifugation at 1000 rpm for 5 min. After that, cold RIPA lysis buffer was added to expose the internalized nanoparticles. The supernatant was obtained by centrifugation at 14 000 rpm for 15 min, dispersed in acetone and then analyzed by a fluorescence spectrometer and a BCA Protein Quantification Kit (Jiangsu, China) to determine the concentrations of coumarin-6 and the cellular protein content, respectively.
Cytotoxicity study
The viability of Caco-2 cells was assessed using Cell Counting Kit-8 (CCK-8) assay after treatment with coumaine-6-loaded nanoparticles. In brief, the cells were seeded in 96-well plates at a density of 5 × 103 cells/well in 100 μL culture medium and incubated for 24 h at 37 °C, 5% CO2. Then, the cells were treated with 10 μL of nanoparticles suspensions for 6 h. Following co-incubation with nanoparticles, 10 μL of CCK-8 solution was added to each well and incubated with the cells for another 2 h. Absorbance was determined at 450 nm using a microplate reader (DNM-9602A, Perlong, Beijing, China). The cells without any treatment were used as a control.
Pharmacodynamics and pharmacokinetics in vivo
The in vivo experiments were conducted on normal male Sprague-Dawley rats of 180 ± 20 g. All the experiments complied with the law and ethics of the People’s Republic of China on the use of experimental animals. The rats were divided into five groups randomly with seven of each group and fasted overnight with free access to water supply before the experiments. Prior to ileal segments administration, the rats were anesthetized by intraperitoneal injection of sodium pentobarbital (45 mg/kg) and fixed on the operation table. The following surgical operation was carried out on the basis of early studies (Morishita et al., Citation2007; Liu et al., Citation2013b). The ileum was exposed after the midline laparotomy and then rinsed with saline at 37 °C to remove the intestinal content. After 30 min of recovery, the test insulin-loaded nanoparticles including Pen-NPs, Sec-Pen-NPs and unmodified nanoparticles at an insulin dose of 10 IU/kg, as well as saline, were respectively injected into the selected ileal segments. Following the administration, the abdominal incision was sutured carefully. One group treated with the same surgical operation as mentioned above was administrated with insulin solution by subcutaneous injection at a dose of 1 IU/kg. Blood samples were taken from the retro-orbital plexus of rats before drug administration and at defined time points after dosing, and centrifuged immediately at 13 000 rpm for 5 min. The obtained plasma was analyzed by using Rat INS (Insulin) ELISA Kits (Hubei, China) and a glucose meter to determine the plasma insulin concentration and the blood glucose levels, respectively.
The relative bioavailability (BA%) of the test nanoparticles by ileal segments administration (i.d.) was evaluated relative to the subcutaneous injection (s.c.) by calculating the area under the plasma insulin concentration versus time curve (AUC) using the following equation:
(4)
where, AUCi.d. and AUCs.c. are the area under the plasma insulin concentration versus time curve by ileal segments administration and subcutaneous injection, respectively. Di.d. and Ds.c. are the dosage of insulin following ileal segments administration and subcutaneous injection, respectively. The hypoglycemic effect was estimated by calculating the area above the blood glucose levels versus time curve (AAC).
Statistical analysis
All data were expressed as means ± SD. The statistical analysis of data was performed using Student’s t-test. The statistical difference was considered significant when the p value is less than 0.05.
Results and discussion
Preparation and characterization of nanoparticles
This study attempted to examine whether nanoparticles co-modified with Sec and CPPs would act as a useful strategy for the oral delivery of insulin. Three different cationic CPPs, including Pen, octaarginine (R8) and Tat, were used here since they are the most extensively utilized CPPs. The N-terminus of CPPs and Sec was all conjugated to stearic acid. It was reported that STR-R8 modified multifunctional envelope-type nano device (MEND) was a useful vehicle for the delivery of nucleic acids. The modification was easily achieved by adding STR-R8 to the liposomal solution, and then the stearyl moiety would insert into liposomal membranes (Khalil et al., Citation2007). STR-CPPs (R8, Tat, Pen) and STR-Sec used in the present study might insert into nanoparticles by the similar way. The amount of STR-CPPs and/or STR-Sec modification on the surface of nanoparticles was greater than 50%, which was quantified by a BCA Protein Quantification Kit (Jiangsu, China) with the wavelength at 562 nm (date not shown).
The schematic illustration of modified nanoparticles is displayed in . As shown in the figure, a low molecular weight emulsifier (SE) was used for the preparation of modified nanoparticles instead of polyvinyl alcohol (PVA), a commonly used high molecular weight emulsifier. Because the function of CPPs and Sec on the surface of nanoparticles might be shielded due to the steric hindrance effect of PVA (Sahoo et al., Citation2002; Yin et al., Citation2007; Xu et al., Citation2013). As a control group, unmodified nanoparticles were prepared using PVA as the emulsifier. In a recent report, Xu et al. (Citation2013) showed that when low molecular weight emulsifiers were used for the preparation of PLGA-PEG nanoparticles, rapid penetration of nanoparticles across human cervicovaginal mucus was observed. In contrast, nanoparticles prepared by high molecular weight emulsifiers, such as PVA were immobilized in the mucus. The explanation was that the use of high molecular weight emulsifiers would sterically hindered the PEG coatings on the surface of nanoparticles.
Figure 1. Schematic illustration of STR-CPPs and STR-Sec co-modified nanoparticles loaded with Ins-SPC complex. SE represents the low molecular weight emulsifier.
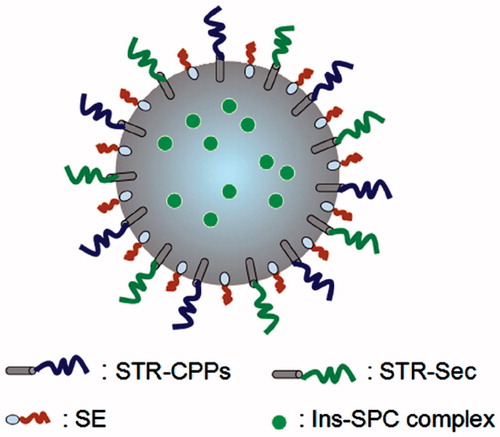
It has been reported that CPPs covalently linked to peptides and proteins were efficiently internalized into cells (Schwarze et al., Citation1999; Liang & Yang, Citation2005). Also, CPPs were conjugated to nanoparticles through a thioether linkage between the C-terminal cysteine of CPPs and nanoparticles for improving the cell permeability of drugs (Koch et al., Citation2005). In these strategies, the chemical conjugation between CPPs and cargoes was required and undesired denaturation of cargoes might occur. However, in our approach, the modification of CPPs and Sec on nanoparticles does not need the covalent conjugation processes, possessing the benefit of avoiding the loss of biological activity of cargoes (Fonseca et al., Citation2009; Yan et al., Citation2012). The electrostatic interaction was caused by the negatively charged nanoparticles and the positively charged cationic CPPs. Moreover, the introduction of stearic acid in the N-terminus of CPPs and Sec would further improve the hydrophobic interactions with nanoparticles. The encapsulated Ins-SPC complex was formulated by an anhydrous co-solvent lyophilization method in order to improve drug liposolubility and thus the encapsulation efficiency (Cui et al., Citation2006). The amounts of residual DMSO in all Ins-SPC complexes were less than 0.0050%, which was far lower than the maximum allowable limits of residual DMSO (0.5%) in the active pharmaceutical ingredients and excipients in accordance with Chinese Pharmacopoeia 2010 edition.
The representative transmission electron microscopy (TEM) images of Pen-NPs and Sec-Pen-NPs demonstrated that the nanoparticles had regular spherical shapes and similar particle size in aqueous solution (). The other test nanoparticles were also spherical in shape and similar to Pen-NPs and Sec-Pen-NPs. The characteristics of nanoparticles (including particle size, zeta potential, encapsulation efficiency and drug loading) are presented in and . For coumarin-6-loaded nanoparticles (), the mean diameters of Sec-R8-NPs, Sec-Tat-NPs and Sec-Pen-NPs were between 140 nm and 160 nm, which were slightly higher than those of R8-NPs, Tat-NPs and Pen-NPs (120 nm–140 nm), respectively. The zeta potential has been used as an index to evaluate surface modification of the two kinds of peptides on nanoparticles. The zeta potential of unmodified nanoparticles loading coumarin-6 was −7.7 ± 1.0 mV. However, after the modification of CPPs and/or Sec, the zeta potential reversed to positive charge, ranging from 30 to 40 mV, due to the positively charged amino acids of CPPs. The encapsulation efficiency and drug loading of coumarin-6-loaded nanoparticles were above 70 and 0.015%. Similar observations were obtained for insulin-loaded nanoparticles (). The mean diameters of Sec-Pen-NPs and Pen-NPs exhibited around 147.5 nm and 163.1 nm, respectively. Unmodified nanoparticles displayed the negative surface charge (−15.6 ± 2.3 mV). In contrast, Pen-NPs and Sec-Pen-NPs were positively charged between 20 and 30 mV. The encapsulation efficiency and drug loading of insulin-loaded nanoparticles were around 60 and 2.5%. As shown in and , the polydispersity indexes (PDI) of particle size of all the formulations were less than 0.3, indicating the uniform size distribution of nanoparticles.
Figure 2. Transmission electron microscopy (TEM) images of Pen-NPs (A) and Sec-Pen-NPs (B). The scale bar represents 200 nm.
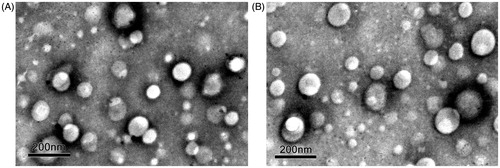
Table 2. Characteristics of coumarin-6 (C6)-loaded nanoparticles (mean ± SD, n = 3).
Table 3. Characteristics of insulin-loaded nanoparticles (mean ± SD, n = 3).
In vitro leakage study of coumarin-6
Fluorescence techniques are widely utilized to track the cellular uptake and intracellular fate of nanoparticles (Du et al., Citation2013). In this study, coumarin-6 was selected as the representative fluorescence probe for assessing in vitro behavior of cellular uptake and transport of nanoparticles. Coumarin-6 was released less than 2% from nanoparticles in both pH 7.4 and pH 4.0 PBS mediums for 12 h (date not shown), implying that almost all of coumarin-6 were still encapsulated in nanoparticles during the in vitro studies. Therefore, it was certain that coumarin-6 would serve as an effective fluorescence probe for the cell association studies.
In vitro release study of insulin
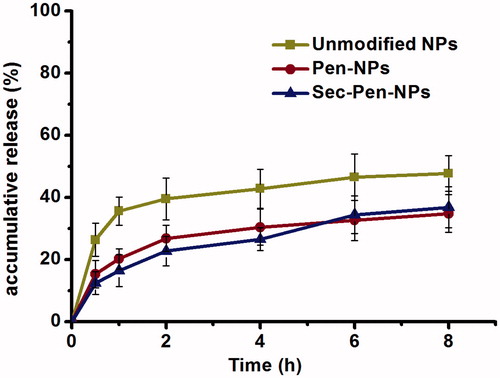
shows the release profiles of insulin from nanoparticles in simulated intestinal medium (PBS, pH 6.8) without trypsin. As seen in , insulin was released about 50% from unmodified nanoparticles for 8 h. The amount of insulin released from Pen-NPs and Sec-Pen-NPs was around 35% for 8 h.
Figure 3. In vitro release of insulin from insulin-loaded nanoparticles (unmodified NPs, Pen-NPs, Sec-Pen-NPs) in simulated intestinal medium (PBS, pH 6.8) without trypsin at defined intervals. Each data point was expressed as the mean ± SD (n = 3).
Enzymatic stability study
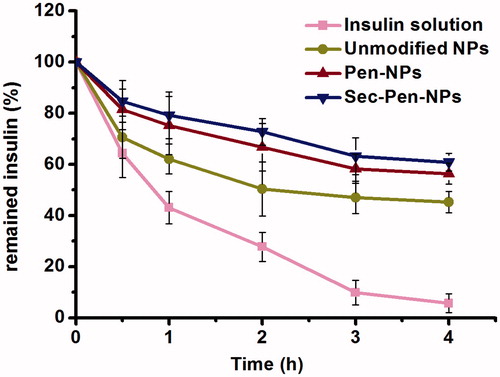
illustrates the percentage of remained insulin after degraded by trypsin in simulated intestinal medium (PBS, pH 6.8). For the control group, about 45% of insulin was degraded at 1 h. And only less than 10% of insulin was remained at 4 h. In contrast, slower degradation rate was found for the test nanoparticles. After 4-h incubation with trypsin, the percentage of remained insulin for unmodified nanoparticles, Pen-NPs and Sec-Pen-NPs was 45.3, 56.4 and 60.9%, respectively. The results suggested that nanoparticles possess the ability to improve the stability of encapsulated drugs.
Figure 4. Remained insulin of insulin solution and insulin-loaded nanoparticles (unmodified NPs, Pen-NPs, Sec-Pen-NPs) after incubation in simulated intestinal medium (PBS, pH 6.8) with trypsin at defined intervals. Each data point was expressed as the mean ± SD (n = 3).
Transcellular transport study
To investigate the effects of CPPs and Sec modification on transcellular transport of nanoparticles, in vitro transport experiments on Caco-2 cell monolayers of modified nanoparticles were carried out. The negligible change of TEER values during the test time demonstrated that the integrity of the cell monolayers was not affected by the addition of the test nanoparticles (data not shown). and show the time profiles of transcellular permeation and the apparent permeability coefficient (Papp) of all the formulations, respectively. The cumulative permeation percentage of all the modified nanoparticles was greater than that of unmodified nanoparticles. The Papp values of R8-NPs, Tat-NPs and Pen-NPs increased to 1.75-times, 1.65-times and 2.15-times compared with unmodified nanoparticles. Among the nanoparticles modified with only CPPs, Pen-NPs displayed the highest cellular transport efficiency. Nanoparticles co-modified with Sec and CPPs (R8, Tat, Pen) produced higher cellular transport efficiency compared to CPPs (R8, Tat, Pen) modified nanoparticles, respectively. The Papp values of Sec-R8-NPs, Sec-Tat-NPs and Sec-Pen-NPs was 1.09-times, 1.12-times and 2.03-times greater than that of R8-NPs, Tat-NPs and Pen-NPs, respectively. Additionally, the Papp values of Sec-R8-NPs, Sec-Tat-NPs and Sec-Pen-NPs was 1.92-times, 1.84-times and 4.36-times increased in comparison with unmodified nanoparticles. The co-modification of Sec and Pen enhanced the cell permeability of nanoparticles the most effectively.
Figure 5. Transcellular transport of coumarin-6-loaded nanoparticles through Caco-2 cell monolayers at defined intervals. Each data point was expressed as the mean ± SD (n = 3).
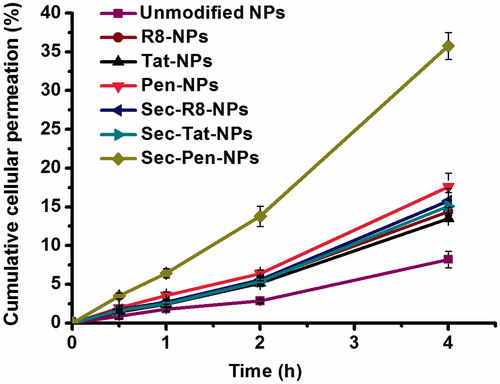
Table 4. Papp of coumarin-6-loaded nanoparticles across the Caco-2 cell monolayers (mean ± SD, n = 3).
Cellular uptake study
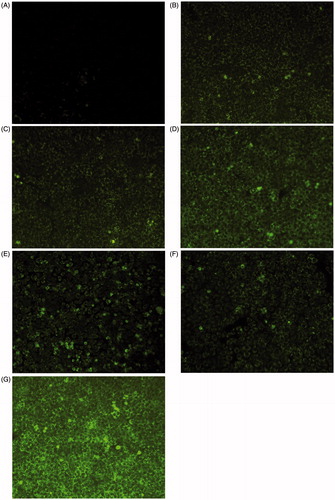
In vitro cellular uptake of modified nanoparticles by Caco-2 cells was next investigated using fluorescence microscopy and fluorescence spectrometer. As seen in , green fluorescence signal was detected in the cells incubated with modified nanoparticles for 2 h. However, a little green fluorescence signal was observed after treatment with unmodified nanoparticles. It was obvious that the Sec-Pen-NPs group exhibited stronger green fluorescence intensity than other modified nanoparticles groups.
Figure 6. Fluorescence microscopy images of Caco-2 cell monolayer incubated with coumarin-6-loaded nanoparticles: unmodified NPs (A), R8-NPs (B), Tat-NPs (C), Pen-NPs (D), Sec-R8-NPs (E), Sec-Tat-NPs (F), Sec-Pen-NPs (G) for 2 h at 37 °C.
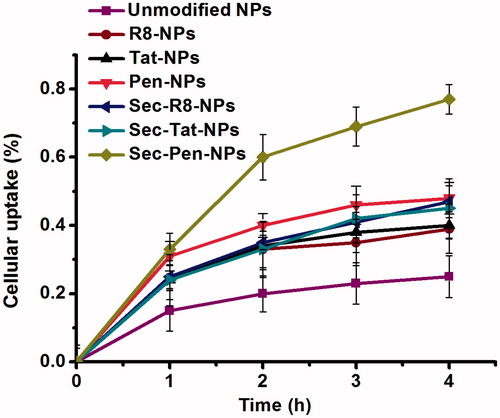
illustrates the time profiles of cellular uptake analyzed by fluorescence spectrometer. R8-NPs, Tat-NPs and Pen-NPs enhanced the cellular uptake by 1.56-times, 1.80-times and 1.92-times compared with unmodified nanoparticles at 4 h. Among CPPs modified nanoparticles, Pen-NPs showed the strongest internalization ability. The cellular uptake amount of Sec-R8-NPs, Sec-Tat-NPs and Sec-Pen-NPs was 1.23-times, 1.15-times and 1.61-times higher than that of R8-NPs, Tat-NPs and Pen-NPs at 4 h, respectively. Obviously, among all the formulations, Sec-Pen-NPs had the highest cellular uptake efficiency.
Figure 7. Cellular uptake of coumarin-6-loaded nanoparticles by Caco-2 cells at defined intervals. Each data point was expressed as the mean ± SD (n = 3).
The cellular uptake study suggested that R8 and Tat showed similar levels of enhancing internalization efficacy of nanoparticles, whereas, Pen exhibited stronger ability to improve cellular uptake than R8 and Tat. The observed differences may be attributed to distinct properties between CPPs. It has been demonstrated that Tat and oligoarginine peptides shared similar modes of internalization (Futaki et al., Citation2001; Suzuki et al., Citation2002; Futaki, Citation2005). However, Pen might utilize some distinct pathways to uptake. As described by Nakase et al. (Citation2004), the uptake of R8 by HeLa cells occurred in a macropinocytosis mode, whereas a distinct pathway for the uptake of Pen was observed.
According to the results obtained from the in vitro cellular uptake and transport studies, Sec-Pen-NPs showed much higher cellular uptake and transport than Pen-NPs. The higher cellular transport of Sec-Pen-NPs was in agreement with their higher cellular uptake. Thus, enhanced cellular transport of Sec-Pen-NPs seemed to be a result of their increased cellular uptake. It seems likely that there is a synergetic effect between Sec and Pen for the improvement of internalization ability. Therefore, the results suggested the improvement of internalization ability by Sec might be one of the main reasons for the increased transcellular transport of Sec-Pen-NPs compared with Pen-NPs. Meanwhile, in the case of Sec-R8-NPs and Sec-Tat-NPs, the contribution of Sec co-modification to the improvement of cellular uptake and transport was not obvious as in the case of Sec-Pen-NPs. This result is consistent with Edmond Dupont’s findings that when tested on MDCK monolayers, SecTat polypeptide could not cross the MDCK epithelium, whereas SecPen polypeptide was capable of transcellular transport across the epithelial layer (Dupont et al., Citation2007). It was indicated that CPPs properties had a major impact on the transcellular property of the chimerical peptide of Sec and CPPs. The transcellular property of SecPen polypeptide was largely dependent on the combined two functionalities: internalization driven by Pen and secretion driven by Sec, whereas Sec could not play its secretion function when applied in combination with Tat (Dupont et al., Citation2007). Our results also showed that the increase of transcellular transport of nanoparticles modified together with R8 and Sec, or Tat and Sec, was not significant compared with nanoparticles modified with only R8 or Tat, which suggested that Sec might not play its secretion function when applied in combination with R8 or Tat. In contrast, a significant increase in cellular transport of nanoparticles was observed when Sec was used together with Pen. This suggested that it could not exclude the possibility that Sec would also favor the step of exocytosis for Sec-Pen-NPs, which need to confirm in the further studies.
Cytotoxicity study
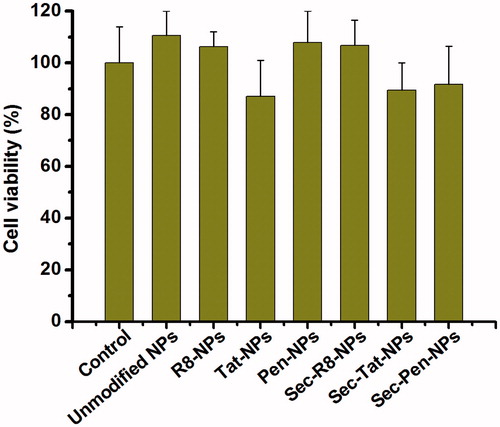
The in vitro cytotoxicity study of modified nanoparticles was investigated on Caco-2 cells. The cells’ viability was assessed using CCK-8 assay to determine whether modified nanoparticles showed toxic effects at the concentration tested. displays the cytotoxicity of all the formulations after incubation with the cells for 6 h. The cells without any treatment were used as a control. As shown in , no apparent cytotoxicity was detected compared to the control at the concentration tested. Although Tat-NPs and Sec-Tat-NPs appeared to cause slightly greater toxic effects than other test nanoparticles, the viability of the cells was all more than 85%. The results indicated that the test modified nanoparticles showed negligible toxic effects on the cells’ viability. Therefore, enhanced cellular uptake and transport of modified nanoparticles in Caco-2 cells was unlikely a consequence of nanoparticles-induced cell death.
Figure 8. CCK-8 assays of cell viability after incubation of coumarin-6-loaded nanoparticles with Caco-2 cells for 2 h. Each data point was expressed as the mean ± SD (n = 3).
Pharmacodynamics and pharmacokinetics in vivo
In the present study, the pharmacokinetic and pharmacodynamic studies were further conducted on normal male Sprague-Dawley rats by ileal segments administration to evaluate the efficacy of modified nanoparticles loaded with insulin. According to the results of in vitro studies, Sec-Pen-NPs and Pen-NPs showed higher cellular uptake and transport efficiency than other modified nanoparticles. Hence, the two modified nanoparticles were used for further in vivo studies by loading insulin.
shows changes in the plasma insulin concentration versus time following ileal segments administration of saline, Pen-NPs, Sec-Pen-NPs, unmodified nanoparticles and s.c. administration of insulin solution. When unmodified nanoparticles were administered, the plasma insulin concentration increased only slightly with a maximum insulin concentration of 30.10 ± 10.67 ng/mL at 4 h. However, the plasma concentrations of insulin in Pen-NPs and Sec-Pen-NPs gradually increased to the peak concentration of 53.92 ± 9.46 ng/mL and 90.23 ± 33.31 ng/mL during 4 h after ileal segments administration, respectively. summarizes the pharmacokinetic and pharmacodynamic parameters of different formulations. The relative bioavailability (BA%) of insulin for Pen-NPs (11.17 ± 2.05%) and Sec-Pen-NPs (19.10 ± 2.58%) were 1.86-times (p < 0.05) and 3.18-times (p < 0.01) higher compared with unmodified nanoparticles (6.01 ± 2.07%). Moreover, the AUC values of Sec-Pen-NPs showed a significant difference compared with that of Pen-NPs (p < 0.05). The relative bioavailability of insulin for Sec-Pen-NPs was 1.71-times higher than Pen-NPs.
Figure 9. Plasma insulin concentration in rats after ileal segments administration of saline, insulin-loaded nanoparticles (unmodified NPs, Pen-NPs, Sec-Pen-NPs) at a dose of 10 IU/kg and s.c. administration of insulin solution (1 IU/kg). Each data point was expressed as the mean ± SD (n = 7).
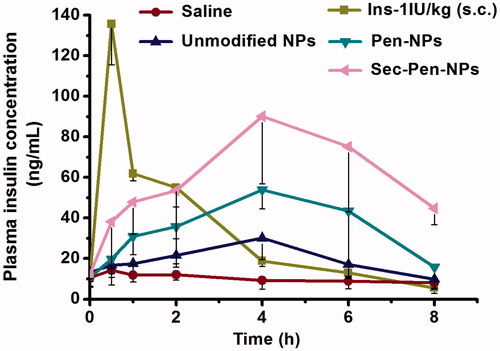
Table 5. Pharmacokinetic and pharmacodynamic parameters in rats after ileal segments administration of insulin loaded nanoparticles (Unmodified NPs, Pen-NPs, Sec-Pen-NPs) and s.c. administration of insulin solution (mean ± SD, n = 7).
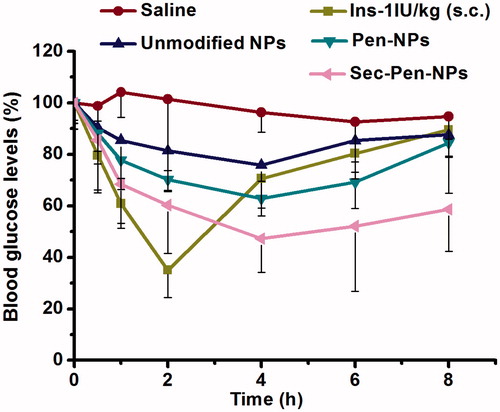
The blood glucose levels following administration of different formulations are displayed in . For the unmodified nanoparticles group, blood glucose levels were reduced to the minimum value of 75.77 ± 6.28% at 4 h, which showed a slight hypoglycemic effect in contrast to the saline group. However, ileal segments administration of Pen-NPs and Sec-Pen-NPs decreased blood glucose to the lowest levels of 62.89 ± 3.79% and 47.28 ± 13.14% at 4 h, respectively. Thus, the two modified nanoparticles displayed stronger hypoglycemic effects compared with unmodified nanoparticles, especially for Sec-Pen-NPs. As shown in , the pharmacological bioavailability (PA%) of insulin for Pen-NPs (9.18 ± 1.32%) and Sec-Pen-NPs (14.72 ± 2.73%) were 1.64-times (p < 0.05) and 2.63-times (p < 0.01) greater than that of unmodified nanoparticles (5.61 ± 1.59%). Moreover, a significant difference in AAC values was found between Pen-NPs and Sec-Pen-NPs (p < 0.05). The pharmacological bioavailability of insulin for Sec-Pen-NPs increased to 1.60-times compared with that of Pen-NPs.
Figure 10. Blood glucose levels in rats after ileal segments administration of saline, insulin-loaded nanoparticles (unmodified NPs, Pen-NPs, Sec-Pen-NPs) and s.c. administration of insulin solution. Each data point was expressed as the mean ± SD (n = 7).
Cell-penetrating peptides have showed remarkable ability to transport a wide variety of therapeutic macromolecules into the interior of cells owing to their high internalization efficiency and potential for structural modification (Patel et al., Citation2009; Desai et al., Citation2010; Lee et al., Citation2011; Mussbach et al., Citation2011). The cellular uptake mechanisms of CPPs have been widely studied by numerous investigations in recent years. It is generally believed that CPPs can utilize multiple pathways of internalization to enter cells, including direct translocation across the plasma membrane, clathrin-mediated endocytosis, caveolae-mediated endocytosis, clathrin/caveolae-independent endocytosis and macropinocytosis (Fonseca et al., Citation2009; Wang et al., Citation2014). But the precise uptake mechanisms of CPPs remain debated, which can be dependent on the properties of CPPs and cargoes, the CPP concentration and cell type (Koren & Torchilin, Citation2012).
Besides, CPPs have been harnessed to traverse epithelial barriers for transepithelial drug delivery (Foged & Nielsen, Citation2008; Koren & Torchilin, Citation2012). Several studies have indicated that CPPs could be applied for enhanced intestinal insulin absorption. In a study published by Morishita et al. (Citation2007), the administration of a physical mixture containing insulin and oligoarginine with intermolecular interactions dramatically enhanced intestinal absorption of insulin. In another study, it was reported that insulin-loaded solid lipid nanoparticles (SLNs) modified with stearic acid-octaarginine (STR-R8) showed a significant hypoglycemic effect compared with unmodified SLNs (Zhang et al., Citation2012). Additionally, Liu et al. (Citation2013b) devised a biodegradable nanoparticle drug carrier modified with oligoarginine (R8) for the delivery of insulin. The modification of R8 was conducted by being covalently linked to the free ends of PEG. The nanoparticles modified with D-R8 and L-R8 promoted the intestinal insulin absorption by 4.4- and 3.2-fold compared with unmodified nanoparticles, respectively.
Here, we designed a strategy using CPPs and Sec with N-terminal stearylation co-modified nanoparticles in order to further increase intestinal absorption of peptide and protein drugs. This nanoparticle-based drug carrier has the advantages of protecting peptide and protein drugs from degradation by enzymes and increasing their retention time in the gastrointestinal tract. On the other hand, as CPPs and Sec may facilitate cargoes go in and out of cells, transcellular transport of insulin in intestinal enterocytes may be improved by using the two kinds of peptides together. In vitro transcellular permeation studies showed Sec-Pen-NPs had much higher transport efficiency than Pen-NPs. The finding was further validated by the pharmacokinetic and pharmacodynamic studies, which demonstrated that Sec-Pen-NPs displayed stronger enhancing effects on ileal insulin absorption than Pen-NPs. Taken together, above results supported that this strategy can effectively improve intestinal insulin absorption.
Conclusion
In summary, Pen enhanced cellular uptake and transport of nanoparticles more effectively than R8 and Tat, especially when used together with Sec. In the case of R8- or Tat-modified nanoparticles, the co-modification of Sec contributed little to the improvement of cellular uptake and transport. In comparison with unmodified nanoparticles, Pen-NPs and Sec-Pen-NPs loaded with insulin were shown to improve ileal insulin absorption by 1.86- and 3.18-times, respectively. Also, stronger hypoglycemic effects were observed, especially for Sec-Pen-NPs. This report provides insight into the use of Sec and Pen to delivery peptide and protein drugs.
Declaration of interest
The authors report no conflicts of interest.
This work was supported by the National Natural Science Foundation of China (Grant No. 81260482 and 30960466) and the National High Technology Research and Development Program of China (Grant No. 2014AA020539).
References
- Bernkop-Schnurch A. (1998). The use of inhibitory agents to overcome the enzymatic barrier to perorally administered therapeutic peptides and proteins. J Control Release 52:1–16
- Bernkop-Schnurch A, Schmitz T. (2007). Presystemic metabolism of orally administered peptide drugs and strategies to overcome it. Curr Drug Metab 8:509–17
- Cui F, Shi K, Zhang L, et al. (2006). Biodegradable nanoparticles loaded with insulin-phospholipid complex for oral delivery: preparation, in vitro characterization and in vivo evaluation. J Control Release 114:242–50
- des Rieux A, Fievez V, Garinot M, et al. (2006). Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J Control Release 116:1–27
- Desai P, Patlolla RR, Singh M. (2010). Interaction of nanoparticles and cell-penetrating peptides with skin for transdermal drug delivery. Mol Membr Biol 27:247–59
- Du W, Fan Y, Zheng N, et al. (2013). Transferrin receptor specific nanocarriers conjugated with functional 7 peptide for oral drug delivery. Biomaterials 34:794–806
- Dupont E, Prochiantz A, Joliot A. (2007). Identification of a signal peptide for unconventional secretion. J Biol Chem 282:8994–9000
- Fan T, Chen C, Guo H, et al. (2014). Design and evaluation of solid lipid nanoparticles modified with peptide ligand for oral delivery of protein drugs. Eur J Pharm Biopharm 88:518–28
- Farkhani SM, Valizadeh A, Karami H, et al. (2014). Cell penetrating peptides: efficient vectors for delivery of nanoparticles, nanocarriers, therapeutic and diagnostic molecules. Peptides 57:78–94
- Foged C, Nielsen HM. (2008). Cell-penetrating peptides for drug delivery across membrane barriers. Expert Opin Drug Deliv 5:105–17
- Fonseca S, Pereira MP, Kelley SO. (2009). Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv Drug Deliv Rev 61:953–64
- Futaki S. (2005). Membrane-permeable arginine-rich peptides and the translocation mechanisms. Adv Drug Deliv Rev 57:547–58
- Futaki S, Suzuki T, Ohashi W, et al. (2001). Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem 276:5836–40
- Gamboa JM, Leong KW. (2013). In vitro and in vivo models for the study of oral delivery of nanoparticles. Adv Drug Deliv Rev 65:800–10
- Goldberg M, Gomez-Orellana I. (2003). Challenges for the oral delivery of macromolecules. Nat Rev Drug Discov 2:289–95
- Gupta V, Hwang BH, Doshi N, et al. (2013). A permeation enhancer for increasing transport of therapeutic macromolecules across the intestine. J Control Release 172:541–9
- Khalil IA, Kogure K, Futaki S, et al. (2007). Octaarginine-modified multifunctional envelope-type nanoparticles for gene delivery. Gene Ther 14:682–9
- Koch AM, Reynolds F, Merkle HP, et al. (2005). Transport of surface-modified nanoparticles through cell monolayers. Chembiochem 6:337–45
- Koren E, Torchilin VP. (2012). Cell-penetrating peptides: breaking through to the other side. Trends Mol Med 18:385–93
- Kumari A, Yadav SK, Yadav SC. (2010). Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces 75:1–18
- Lee JY, Choi YS, Suh JC, et al. (2011). Cell-penetrating chitosan/doxorubicin/TAT conjugates for efficient cancer therapy. Int J Cancer 128:2470–80
- Liang JF, Yang VC. (2005). Insulin-cell penetrating peptide hybrids with improved intestinal absorption efficiency. Biochem Biophys Res Commun 335:734–8
- Liu BR, Liou JS, Chen YJ, et al. (2013a). Delivery of nucleic acids, proteins, and nanoparticles by arginine-rich cell-penetrating peptides in rotifers. Mar Biotechnol 15:584–95
- Liu X, Liu C, Zhang W, et al. (2013b). Oligoarginine-modified biodegradable nanoparticles improve the intestinal absorption of insulin. Int J Pharm 448:159–67
- Mahato RI, Narang AS, Thoma L, et al. (2003). Emerging trends in oral delivery of peptide and protein drugs. Crit Rev Ther Drug 20:153–214
- Morishita M, Kamei N, Ehara J, et al. (2007). A novel approach using functional peptides for efficient intestinal absorption of insulin. J Control Release 118:177–84
- Mussbach F, Franke M, Zoch A, et al. (2011). Transduction of peptides and proteins into live cells by cell penetrating peptides. J Cell Biochem 112:3824–33
- Nakase I, Niwa M, Takeuchi T, et al. (2004). Cellular uptake of arginine-rich peptides: roles for macropinocytosis and actin rearrangement. Mol Ther 10:1011–22
- Panyam J, Labhasetwar V. (2003). Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev 55:329–47
- Patel L, Wang J, Kim KJ, et al. (2009). Conjugation with cationic cell-penetrating peptide increases pulmonary absorption of insulin. Mol Pharm 6:492–503
- Patel L, Zaro JL, Shen WC. (2007). Cell penetrating peptides: intracellular pathways and pharmaceutical perspectives. Pharm Res 24:1977–92
- Renukuntla J, Vadlapudi AD, Patel A, et al. (2013). Approaches for enhancing oral bioavailability of peptides and proteins. Int J Pharm 447:75–93
- Ruan G, Agrawal A, Marcus AI, et al. (2007). Imaging and tracking of tat peptide-conjugated quantum dots in living cells: new insights into nanoparticle uptake, intracellular transport, and vesicle shedding. J Am Chem Soc 129:14759–66
- Sahoo SK, Panyam J, Prabha S, et al. (2002). Residual polyvinyl alcohol associated with poly(d,l-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J Control Release 82:105–14
- Sawant RM, Hurley JP, Salmaso S, et al. (2006). “SMART” drug delivery systems: double-targeted pH-responsive pharmaceutical nanocarriers. Bioconjug Chem 17:943–9
- Schwarze S, Ho A, Vocero-Akbani A, et al. (1999). In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 285:1569–72
- Sethuraman VA, Lee M, Bae YH. (2008). A biodegradable pH-sensitive micelle system for targeting acidic solid tumors. Pharm Res 25:657–66
- Sood A, Panchagnula R. (2001). Peroral route: an opportunity for protein and peptide drug delivery. Chem Rev 101:3275–303
- Stewart KM, Horton KL, Kelley SO. (2008). Cell-penetrating peptides as delivery vehicles for biology and medicine. Org Biomol Chem 6:2242–55
- Su FY, Lin KJ, Sonaje K, et al. (2012). Protease inhibition and absorption enhancement by functional nanoparticles for effective oral insulin delivery. Biomaterials 33:2801–11
- Suzuki T, Futaki S, Niwa M, et al. (2002). Possible existence of common internalization mechanisms among arginine-rich peptides. J Biol Chem 277:2437–43
- Torchilin VP. (2008). Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv Drug Deliv Rev 60:548–58
- Wang F, Wang Y, Zhang X, et al. (2014). Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. J Control Release 174:126–36
- Xu Q, Boylan NJ, Cai S, et al. (2013). Scalable method to produce biodegradable nanoparticles that rapidly penetrate human mucus. J Control Release 170:279–86
- Yan L, Wang H, Jiang Y, et al. (2012). Cell-penetrating peptide-modified PLGA nanoparticles for enhanced nose-to-brain macromolecular delivery. Macromol Res 21:435–41
- Yin Y, Chen D, Qiao M, et al. (2007). Lectin-conjugated PLGA nanoparticles loaded with thymopentin: ex vivo bioadhesion and in vivo biodistribution. J Control Release 123:27–38
- Zhang ZH, Zhang YL, Zhou JP, et al. (2012). Solid lipid nanoparticles modified with stearic acid-octaarginine for oral administration of insulin. Int J Nanomed 7:3333–9