
Abstract
Colon is an ideal absorptive site for oral protein and peptide drug (insulin), and yet it poses multiple barriers against the drug absorption, such as the barriers against the drug diffusion from colon lumen toward the absorptive mucosa and permeation across colon epithelium. In this study, modified nanoparticles (Tat-CS-NPs) with cell-penetrating peptide (Tat) and amphipathic chitosan derivative (a-CS) were used as carriers to improve colonic absorption of the drug. With a-CS as emulsifier and poly(lactic-co-glycolic acid) as matrix material, Tat-CS-NPs were prepared and evaluated in vitro/vivo. Using Caco-2 cell monolayers to imitate the colonic epithelial cells, it was found that the cellular uptake amount and transcellular transportation performance of Tat-CS-NPs were much enhanced compared with those of CS-NPs and PVA-NPs. The efficacy evaluation on diabetic rat models demonstrated that the hypoglycemic effect of Tat-CS-NPs loaded with insulin was 6.89 times higher than that of PVA-NPs, and 1.79 times higher than that of CS-NPs. By gavage of [99mTc] isotope labeled Tat-CS-NPs in mini-pigs and single-photon emission computed tomography (SPECT) on pigs' gastrointestinal tract, it was further proved that the nanoparticles could reach colon and produce pharmacological effect. In conclusion, Tat-CS-NPs as vehicles for colon-specific drug delivery may be an efficient approach to improve oral bioavailability of protein and peptide drugs.
Introduction
Diabetes mellitus, a group of metabolic diseases characterized by hyperglycemia over a prolonged period, has resulted in numerous deaths worldwide in the past years (Wild et al., Citation2004). Multiple daily injections of insulin, which bring a lot of inconvenience and huge pain, are by far the first choice for insulin-dependent diabetes mellitus (IDDM or Type I diabetes) patients (Skyler, Citation2007; Zhang et al., Citation2010). In recent years, extravascular administration of insulin, including oral, pulmonary, nasal, ocular and transdermal routes, etc. has attracted more and more attention from pharmaceutical scientists (Trehan & Ali, Citation1998; Khafagy et al., Citation2007). Oral dosing, with broad acceptance and cost-effectiveness, has been the focus of most concern to fulfill the expectation of a needle-free insulin treatment (Iyer et al., Citation2010; Bakhru et al., Citation2013).
Nevertheless, as a protein and peptide drug, insulin in its native form has an oral bioavailability ranging from 0.05% to 0.5% producing no clinical effect (Arbit, Citation2004; Peppas & Kavimandan, Citation2006). Two facts are responsible for that: its proneness to acid and enzyme degradation in gastrointestinal tract (GIT) and the barriers against its biomembrane penetration which limits absorption of the drug (Ensign et al., Citation2012; Rekha & Sharma, Citation2013). Compared with other sites in GIT, colon has a physiological environment which assists protein and peptide drugs absorption due to lower density and activity of colonic protease, neutral pH value within and enabling long-time drug residence (Haupt & Rubinstein, Citation2002; Sinha et al., Citation2007; Patel, Citation2011). Hence, colon may be chosen as an ideal absorptive site for oral protein and peptide drugs. However, there are still some barriers against absorption of the drug in colon, for example, the barriers against the drug diffusion from colon lumen toward the absorptive mucosa and permeation across colon epithelium. Colon exhibits a wide lumen that limits a close contact between the absorptive epithelium and the contents (Varum et al., Citation2008), and the contents in colon are quite sticky and hardly flowable which impede the drug diffusion and attachment to mucosa (Varum et al., Citation2008; Maroni et al., Citation2012). Moreover, colonic absorption of proteins and peptides has remained to be a great challenge owing to the low permeability across the colonic epithelium membrane caused by inherent high molecular weight and hydrophilicity of the drug, and the narrow tight junctions may impose restrictions on its paracellular permeability (Maroni et al., Citation2012). As a result, colonic absorption of the drug should be still rather poor (Varum et al., Citation2008; Maroni et al., Citation2012; Patel, Citation2013). In the present study, modified nanoparticles (Tat-CS-NPs) with cell-penetrating peptides (CPPs) (Tat) and amphipathic chitosan derivative (a-CS) were devised as vehicles of the drug to overcome above bottlenecks in oral colon delivery.
Chitosan (CS) is a polycationic polysaccharide derived from natural chitin by partial deacetylation. As a biodegradable, biocompatible, non-toxic and non-immunogenic polymer, it was widely used in cosmetology, regenerative medicine and pharmaceutics (Hejazi & Amiji, Citation2003; Shi et al., Citation2006; Bernkop-Schnürch & Dünnhaupt, Citation2012). Many studies confirmed that chitosan and its synthetic derivatives are endowed with good bioadhesive ability, which may be applied to colonic bioadhesive drug delivery systems (Zhang et al., Citation2002; Yin et al., Citation2009; Shelma & Sharma, Citation2011). Colonic mucoadhesion may facilitate close contact between the nanoparticles and colonic mucosa and help decrease the diffusion paths of the nanoparticles to the mucous surface. Thus, it may lengthen retention time of the nanoparticles in colon. Furthermore, due to effect of steric hindrance, chitosan may protect CPPs from being degraded thus allow CPPs to bring into play its excellent transcellular transportation capability.
CPP is a short-chain polypeptide with a length of less than 30 amino acids generally. Many studies showed that some CPPs, such as Tat, oligoarginine, penetratin and transportan, have the potential to convey cargos, such as nanoparticles, liposomes and micelles, into various cells (Kaplan et al., Citation2005; Torchilin, Citation2008; Foged & Nielsen, Citation2008; Milletti, Citation2012 Liu et al., Citation2013; Wang et al., Citation2014). In addition, it was applied in transepithelial drug delivery (e.g. transdermal delivery and nasal administration), from which good effects were acquired (Tréhin et al., Citation2004; Santra et al., Citation2005; Chauhan et al., Citation2007; Johnson et al., Citation2011; Koren & Torchilin, Citation2012; Khafagy & Morishita, Citation2012; Reissmann, Citation2014). In this study, CPP was applied in colonic transepithelial drug delivery system in order to improve the permeability of protein and peptide drugs.
Tat-CS-NPs were prepared and evaluated in vitro/vivo in this study. A-CS was adopted to give nanoparticles good colonic bioadhesive ability conquering the diffusion barrier and Tat was used to increase transepithelial transportation of nanoparticles in colonic epithelial cells overcoming the penetration barrier. To verify this conception, Tat-CS-NPs were evaluated using Caco-2 cell monolayer models and diabetic rat models, respectively. In addition, SPECT was used to visualize how the nanoparticles travel in mini-pig's GIT at particular intervals.
Materials and methods
Materials and animals
Chitosan (molecular weight 100 kDa, 90% deacetyiation) was kindly donated by Zhejiang Aoxing Biological Technology Co., Ltd. (Zhejiang, China). Porcine insulin (potency 28.7 IU/mg) was purchased from Xuzhou Wanbang Biochemical Pharmaceutical Co., Ltd. (Jiangsu, China). Stearyl-Tat (Ste-Tat) was obtained from GL Biochem (Shanghai) Ltd. (Shanghai, China). Soybean phospholipid (PC ≥ 90%, of injection class) was purchased from Shanghai Taiwei Pharmaceutical Co., Ltd. (Shanghai, China). Lauric aldehyde (analytical reagent, AR) and methyl iodide (AR) were purchased from Xi'a Reagent Co., Ltd. (Sichuan, China). Polyvinyl alcohol (Kuraray PVA-124, molecular weight 105 000) was generously supplied by Guangzhou Dingfengsheng Chemical Technology Co., Ltd. (Guangdong, China). Potassium iodide was obtained from Shanghai Jingchun Chemical Reagent Co., Ltd. (Shanghai, China). Potassium borohydride (PBH) was obtained from Sinopharm Group Co., Ltd. (Shanghai, China). Poly(lactic-co-glycolic acid) (PLGA) (molecular weight 8000, GA/LA ratio 50:50) was obtained from Shandong Institute of Medical Instruments (Shandong, China). Coumarin-6 and streptozotocin were obtained from Sigma Co., Ltd. (St. Louis, MO). Corning transwell® polycarbonate membrane 12-well plates and 96-well plates were purchased from Corning Co., Ltd. (New York, NY). Dialysis bag (molecular weight cut-off 8000–14 000), Hank's balanced salt solution (HBSS), RPMI-1640 culture medium, PBS and TransSerum HQ Fetal Borine Serum were obtained from Solarbio Co., Ltd. (Beijing, China). Ultrafiltration centrifugal tubes were purchased from Millipore Co., Ltd. (100 kD, Millipore, MA). Cell counting kit-8 (CCK-8) was obtained from Boster Biological Technology Co., Ltd. (Hubei, China). RIPA lysis buffer was obtained from Vazyme Biotech Co., Ltd. (Jiangsu, China). Pentobarbital sodium was obtained from Merck KGaA (Darmstadt, Germany). Sodium pertechnetate [99mTc] was purchased from HTA Co., Ltd. (Beijing, China). All other chemicals were of analytical grade or HPLC grade.
Wistar male rats (certification No. 201403240) weighing 200 ± 20 g, 12 weeks old, were supplied by Hunan Experimental Animal Center (Hunan, China). Male Guangxi BA-MA Mini-Pigs (certification No. 201402220) weighing 10 ± 0.5 kg, 5–7 months old, were purchased from Taizhou Taihe Biotechnology Co., Ltd. (Jiangsu, China). All animal experiments complied with the law and ethics of the People's Republic of China on the use of experimental animals.
Synthesis of a-CS (N,N,N-trimethyl-N-dodecyl chitosan)
Lauraldehyde (1.2 ml) was added into the suspension consisting of chitosan (2 g) and methonal (100 ml), and magnetic stirring was conducted on it for 24 h (Zhang et al., Citation2006). PBH (5 ml, 0.1 g/ml) was dropped in and magnetic stirring was carried out for another 24 h. Hydrochloric acid (2 mol/l) was added to neutralize the solution, 300 ml methanol was put in, the precipitate was formed and it was collected through filtering. The precipitate was washed three times using water and methanol and dried by vacuum drier, and N-dodecyl chitosan was gained. Then, 1-methyl-2-pyrrolidinone (80 ml) was used to swell N-dodecyl chitosan (2 g). The suspension was heated to 60 °C and dissolved by the addition of potassium iodide (KI) (5.3 g). Fifteen percent sodium hydroxide (NaOH) (11 ml) and methyliodide (CH3I) (11 ml) were put in the reaction for 1 h, then KI (2.6 g), 15% NaOH (5.5 ml) and CH3I (5.5 ml) were added for reaction for another 45 min. Four hundred milliliters ethanol was poured in to stop the reaction, and the suspension was allowed to stand for sediment. Being centrifuged, the sediment was washed using alcohol, dissolved using proper amount of distilled water and dialyzed in a dialysis bag (14 000 molecular weight cut-off) filled with 15% sodium chloride solution for 3 days and then in the bag with distilled water for another 3 days, each with a dialyzate renewal every 12 h. The liquid in the dialysis bag was centrifuged, and the supernatant obtained was frozen drying by Freeze drier (FD-1A-50, Beijing Bo Kang Experimental Medical Instrument Co., Ltd. Beijing, China). A white cotton-like solid appeared, which was N,N,N-trimethyl-N-dodecyl chitosan.
Characterization of chitosan and N,N,N-trimethyl-N-dodecyl chitosan
Fourier transform infrared spectroscopy (FT-IR) analysis was used to confirm the N-alkyl and N-methyl substitution. FT-IR spectra were recorded using Fourier-transform infrared spectrometer (8400, Shimadzu Corporation, Kyoto, Japan) in KBr disks.
1H NMR spectra were also performed to evaluate the N-alkyl and N-methyl substitution and to determine the degree of trimethyl substitution. NMR was conducted using a nuclear magnetic resonance spectrometer (Bruker AVANCE III 600 MHz, Zurich, Switzerland). Chitosan was dissolved in the mixed solvent of D2O and F3CCOOD, while N,N,N-trimethyl-N-dodecyl chitosan was dissolved in D2O.
Preparation of nanoparticles
Preparation of insulin phospholipid complex
Soybean phospholipid (600 mg) and insulin (40 mg) were placed in a brown glass vial, and dimethyl sulfoxide (DMSO) (15 ml) including glacial acetic acid (0.75 ml) was added (Cui et al., Citation2006). The suspension was stirred in a 30 °C water bath for 1.5 h to enable the solid fully dissolved, and insulin phospholipid complex was obtained after the solution was frozen drying at −55 °C using a freeze-drier.
Preparation of nanoparticles
Nanoparticles were prepared using emulsion-solvent evaporation method. The first step was to prepare O/W emulsion, and then nanoparticles were formed by removing the organic solvents from the emulsion through magnetic stirring at room temperature. In brief, a compound of insulin phospholipid complex (20 mg) and PLGA (25 mg) were dissolved in a mixture (1 ml) of dichloromethane and acetone at the weight ratio of 3:1, as oil phase. Stearyl-Tat (Ste-Tat) (4 mg) was dissolved in distilled water (4 ml) as aqueous phase І. Three milliliters of 1% N,N,N-trimethyl-N-dodecyl chitosan (a-CS) solution was treated as aqueous phase ІІ. The oil phase was dropped into aqueous phase І, and the mixture was shearing for 20 s at 11 000 rpm (High speed shearing, IKN GmbH, Neustadt, Germany). Aqueous phase ІІ was added and the new mixture was shearing for another 20 s at the same speed. The final solution was dispersed with 300 W miniprobe sonography (SCIENTZ-II, Ningbo Scientz Biotecnology Co., Ltd., Zhejiang, China) for 2 min in ice-bath, O/W emulsion was prepared, and then magnetic stirring was carried out to remove the organic solvents at room temperature. Tat-CS-NPs were obtained (schematic diagram shown in ). With the same oil phase and aqueous phase ІІ only, amphipathic chitosan derivative modified nanoparticles (CS-NPs) were prepared in the same way. PVA nanoparticles (PVA-NPs) were prepared as described above using the same oil phase and 3 ml of 1% PVA solution as aqueous phase. Regarding blank nanoparticles (Blank Tat-CS-NPs), they were prepared like Tat-CS-NPs, without insulin phospholipid complex encapsulated. Coumarin-6-loaded NPs (Tat-CS-NPs, CS-NPs, PVA-NPs) were prepared as for their insulin-loaded counterparts, with coumarin-6 replacing insulin phospholipid complex.
Figure 1. Schematic diagram of Tat-CS-NPs.
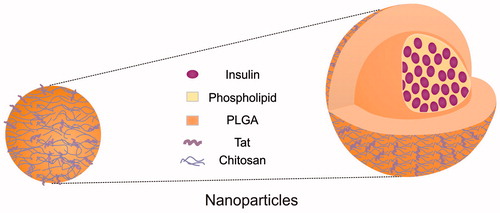
Characterization of nanoparticles
Particle size and zeta potential
The particle size and zeta potential were determined by Malvern Mastersizer (PSA NANO2590, Malvern Instruments Ltd., Malvern, UK). All measurements were performed in triplicate.
Morphology observation
The morphology of Tat-CS-NPs was observed under a transmission electron microscope (TEM, JEM-2100, Japan Electronics Corporation, Tokyo, Japan). The samples were diluted 50-fold with distilled water, and some solution was dropped on the copper screen for volatilization. Then, a drop of 3% phosphotungstic acid solution was used for negative staining. Being dried for 20 min, these nanoparticles were put under the TEM for morphology observation.
Determination of entrapment efficiency and drug loading
Nanoparticles were centrifuged by ultracentrifuge (Optima L-100K, Beckman Coulter Inc., Brea, CA) at 40 000 rpm for 30 min at 10 °C. The supernatant was analyzed for insulin concentration using HPLC (Agilent 1260, Agilent Technologies, Santa Clara, CA). All experiments were performed in triplicate to calculate encapsulation efficiency (EE) and drug loading (DL) by the following equations:
(1)
(2)
In vitro release studies
In vitro release in simulated gastric fluid
The nanoparticles were dispersed in simulated gastric fluid (10 ml, pH 1.2 hydrochloric acid solution without pepsin), and the solution was shaken at 100 rpm by the water bath shaker (Xiaoyang Electric Instrument Factory, Jiangsu, China) at 37 ± 0.5 °C. One milliliter of the solution was taken out at 0.25, 0.5, 0.75, 1, 1.5, 2, 4 and 6 h, respectively, and put into an ultrafiltration centrifugal tube (100 kD molecular weight cut-off) for centrifugation at 3500 rpm for 15 min. The removed volume each time was supplemented with the same amount of fresh medium. The concentration of insulin was determined using HPLC.
In vitro release in simulated intestinal fluid
The nanoparticles were dispersed in simulated intestinal fluid (10 ml, pH 7.4 phosphoric acid buffer solution (PBS) without tripsin), and the solution was shaken at 100 rpm by the water bath shaker at 37 ± 0.5 °C. One milliliter of the solution was taken out at 0.5, 1, 1.5, 2, 4, 6, 8, 10, 12, 24, 36 and 48 h, respectively, and put into an ultrafiltration centrifugal tube (100 kD molecular weight cut-off) for centrifugation at 3500 rpm for 15 min. The removed volume each time was supplemented with the same amount of fresh medium. The concentration of insulin was determined using HPLC.
Cell studies
Cell culture
Human colon carcinoma Caco-2 cells were cultured in 75 cm2 culture flasks with culture medium (Kamei et al., Citation2013). The RPMI-1640 medium contained 10% fetal bovine serum, 1% non-essential amino acids, 1% l-glutamine, 100 mg/ml streptomycin and 100 IU/ml penicillin. The culture flasks were maintained in an incubator at 37 °C, 95% relative humidity, with 5% CO2. When reaching 80% confluence, cells were harvested. Then, they were detached from the culture flasks by trypsinization with 0.25% trypsin containing 0.05 mM ethylenediamine tetraacetic acid, and diluted to a density of 1.0 × 105 cells/cm2. The cell suspensions were seeded onto several transwell® plates, some of which were incubated for 2 days before cytotoxicity studies and the others were incubated for 5 days before uptake studies. For transcellular permeation study, the cell culture density was 1.0 × 105 cells/cm2. Caco-2 cells were grown in the culture medium (DMEM) described above for 21–24 days until the transepithelial electrical resistance (TEER) of them finally achieved constant value (TEER > 500 Ω cm2). TEER values were measured with a Millicell-ERS (Millipore, MA).
Cytotoxicity
The cytotoxic effects of the nanoparticles were assessed using CCK-8 assay (He et al., Citation2013). Caco-2 cells were seeded in a 96-well plate at 5 × 104 cells/cm2 with 200 µl culture medium under 37 °C and 5% CO2. First, cells were incubated with NPs for 2 h (the concentrations of NPs were 0.1, 5 and 10 mg/ml). Then, nanoparticle dispersion in each well was replaced by 200 ml CCK-8 solution, and cells were further incubated for another 2 h. Finally, the absorbance of cell medium in each well was measured at 450 nm via a microplate reader (9602A, Beijing Perlong Medicare Co., Ltd., Beijing, China).
Uptake assay
Coumarin-6-loaded NPs were centrifuged at 40000 rpm for 30 min at 10 °C and the precipitate was dispersed in HBSS. The nanoparticle suspension was filtered through a 0.22-μm filtering film for sterilization. When Caco-2 cells incubated in the plate have reached above 80% confluence, HBSS at 37 °C was used to rinse the cells twice. One milliliter of HBSS was added into each well for incubation for 30 min and then discarded. Then, 0.5 ml of nanoparticle dispersion was put into every well in some plates for 1-h incubation and in other plates for 2-h incubation, respectively, and the residual liquid was discarded. Remaining cells were fully rinsed twice using HBSS at 4 °C, and observed under a fluorescence inversion microscope (Olympus IX71, Tokyo, Japan). Alternatively, 100 µl of RIPA lysis buffer was added into each well which was then oscillated in an ice-bath for 20 min. After cells were detached, acetone was used to dilute and destroy the nanoparticles within, and the fluorescent intensity of coumarin-6 was determined by fluorescence spectrophotometer (LS55, PerkinElmer, CT). The excitation and emission wave length of coumarin-6 was 450 and 502 nm, respectively.
Transcellular permeation study
Coumarin-6-loaded NPs were centrifuged at 40 000 rpm for 30 min at 10 °C, and the precipitate was dispersed in HBSS. The suspension were filtered at 0.22 μm and sterilized for use. Caco-2 cell monolayers (TEER > 500 Ω·cm2) in transwell® plates were also prepared for the study. First, culture medium in the apical and basolateral chambers was replaced with HBSS, and the plate was incubated for 30 min at 37 °C. Then, the Caco-2 cells were rinsed twice with 1 ml HBSS, and incubated with 0.5 ml of coumarin-6-loaded NPs (PVA-NPs, CS-NPs, Tat-CS-NPs) suspension in apical chamber successively and with 1 ml HBSS in basolateral chamber at 37 °C. At the determined time points (0.5, 1, 2 and 4 h), 0.1 ml of samples were taken from the basolateral chamber and equal volume of HBSS was supplemented. After the nanoparticles were destroyed by the addition of acetone, the amount of transported coumarin-6 was determined using fluorescence spectrophotometer and calculated via the standard curve of coumarin-6: A = 664.47C + 8.8494 (r = 0.9996), where A stands for the fluorescence intensity, C represents the concentration.
Pharmacodynamic evaluation
Induction of diabetes
Wistar male rats weighing 200 ± 20 g were fasted overnight prior to the study, but were allowed water ad libitum. Then, diabetes was induced in rats by an intraperitoneal injection of streptozotocin (0.1 mol/l citrate buffer solution, pH 4.5) at the dose of 60 mg/kg (Damgé et al., Citation2007). Blood samples were collected and blood glucose levels were measured using a Roche glucometer (ACCU-CHEK per forma, Basel, Switzerland). Rats whose fasting glycemia were above 16.7 mmol/l 1 week after streptozotocin treatment was considered to be diabetic.
Hypoglycemic effects
Thirty-six diabetic rats were randomly divided into six groups. All were fasted for 8 h but were allowed water ad libitum. After that they were intraperitoneally injected with pentobarbital sodium at the dose of 25 mg/kg for surgical procedure with small incision in their abdomen cavity (Bayat et al., Citation2008). Thereafter, physiological saline was injected into ascending colon portion of the first group of rats, and insulin solution, PVA-NPs, CS-NPs and Tat-CS-NPs were injected into ascending colon portion of the second, third, fourth and fifth group of rats at the dose of 20 IU/kg, respectively. The sixth group was subcutaneously injected with insulin solution at the dose of 1 IU/kg. Blood samples were collected from tail veins of all the rats at 0, 0.5, 1, 2, 4, 6, 8, 12 and 24 h after dosing, and the blood glucose levels were determined. The area under the blood glucose concentration–time curve over 24 h was calculated according to the trapezoidal rule. The pharmacological bioavailability was evaluated by the area above the curve (AAC) of blood glucose for 0–24 h. The total decreases (D%) were calculated using a modification method as follows (Jin et al., Citation2012) :
(3)
Gamma-scintigraphy studies
Radiolabeling procedure of nanoparticles
One hundred milligrams of nanoparticles was dispersed in 5 ml of distilled water forming suspension, to which 0.5 ml of stannous chloride (SnCl2) (5 mg/ml, 0.1 M HCl) and 1.2 ml of sodium pertechnetate [99mTc] (58 mCi) were added (Subramanian et al., Citation2013). The whole suspension was put in a shaker (100 rpm) for incubation for 1 h at room temperature, and centrifuged at 40 000 rpm for 30 min. The clear supernatant was removed, and the precipitation was dispersed in distilled water, which was 99mTc isotope labeled nanoparticle suspension.
Gamma-scintigraphy test
Three Guangxi BA-MA Mini-Pigs (10 ± 0.5 kg) were selected for parallel experiments. They were fasted for 12 h, thereafter gavage of 99mTc isotope labeled nanoparticle suspension was conducted on them. SPECT (Discovery VH, General Electric Co., Ltd., CT) was utilized to visualize the running behaviors of the nanoparticles in their GIT (stomach, small intestine and colon) at 2, 6 and 10 h, respectively.
HPLC analysis of insulin
An HPLC method was adopted for the determination of insulin concentrations in the entrapment efficiency, drug loading, in vitro release studies. HPLC determinations were performed with the Agilent 1260 infinity liquid chromatography system. The mobile phase was a mixture of acetonitrile and 0.2 mol/l anhydrous sodium sulfate solution (adjusted to pH 2.3 with phosphoric acid) at the ratio of 28:72 (v/v). Samples (20 μl) were injected into HPLC system with a flow rate of 1.0 ml/min at a column temperature of 40 °C and the UV detection was performed at 214 nm.
Statistical analysis
One-way analysis of variance (ANOVA) was used to determine significant difference between sample groups. A value of p < 0.05 was considered to be significant difference. All data were expressed as means ± SD.
Results
Characterization of chitosan derivative
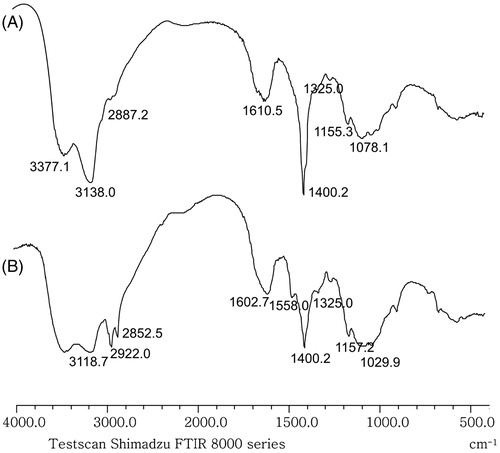
FT-IR spectra of chitosan and N,N,N-trimethyl-N-dodecyl chitosan are shown in . For N,N,N-trimethyl-N-dodecyl chitosan, there were characteristic absorption peaks at about 2922, 2852, 1602, 1558, 1400 cm−1 (). Compared with the spectrum of chitosan (), it is found that peaks at 2852 and 2922 cm−1 in were intensified which suggests the presence of N-alkyl substitution. Meanwhile, new absorption peaks were observed at about 1558 cm−1 in , owing to deformation vibration of NH in NH-alkyl chain.
Figure 2. FT-IR spectra of chitosan and its synthetic derivative. (A) FT-IR spectrum of chitosan and (B) FT-IR spectrum of N,N,N-trimethyl-N-dodecyl chitosan.
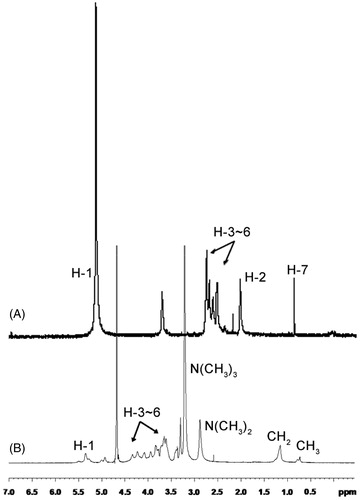
1H NMR spectra of chitosan and N,N,N-trimethyl-N-dodecyl chitosan are shown in . Compared to that of chitosan (), N,N,N-trimethyl-N-dodecyl chitosan had three additional peaks at 3.32, 1.29 and 0.82 ppm in its 1H NMR spectra (). 1H NMR spectrum exhibited the presence of the trimethyl amino group (N(CH3)3) [δ 3.32 (9H, s)], 11 methene groups of N-lauryl group (–NH(CH2)11CH3) [δ 1.29 (22H, m)] and methyl group at N-lauryl tail of (–NH(CH2)11CH3) [δ 0.82 (3H, m)].
Figure 3. 1H NMR spectra of chitosan and its synthetic derivative. (A) 1H NMR spectrum of chitosan and (B) 1H NMR spectrum of N,N,N-trimethyl-N-dodecyl chitosan.
Characterization of nanoparticles
The characteristics of Blank Tat-CS-NPs and drug-loaded NPs (including Tat-CS-NPs, CS-NPs and PVA-NPs) are summarized in . As for particle size, there are small differences between the four nanoparticles. Besides, particle sizes of coumarin-6-loaded NPs were found similar to those of insulin-loaded NPs. exhibits the typical droplet size distribution diagram of Tat-CS-NPs. The average particle size was about 155 nm and most of the sizes were below 100 nm. Moreover, it is apparent that the zeta potential of Tat-CS-NPs was quite distinct from that of PVA-NPs. Tat-CS-NPs had a zeta potential of 53 ± 0.5 mV, while the zeta potential of PVA-NPs was 3.7 ± 0.4 mV. However, EE% and LD% of Tat-CS-NPs were both lower than those of PVA-NPs. EE% and LD% of Tat-CS-NPs were 58.95 ± 5.2% and 1.38 ± 0.35%, respectively, while those of PVA-NPs were 67.21 ± 3.4% and 1.57 ± 0.21%, respectively.
Figure 4. Typical droplet size distribution diagram of Tat-CS-NPs.
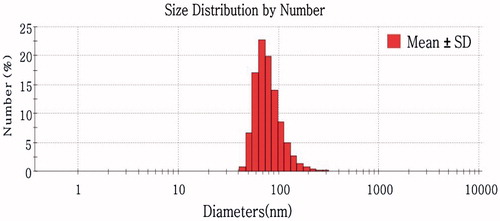
Table 1. Particle size, zeta potential, encapsulation efficiency and drug loading of PVA-NPs, CS-NPs, Tat-CS-NPs and Blank Tat-CS-NPs (Mean ± SD, n = 3).
As can be seen in , the morphology of Tat-CS-NPs was observed under TEM, which turned out to be spherical in shape and uniform.
Figure 5. Transmission electron micrograph of Tat-CS-NPs (×40 000).

In vitro release studies
It can be clearly seen from , in simulated gastric fluid (pH 1.2 hydrochloric acid solution), insulin release of Tat-CS-NPs ascended slightly, reaching 10% at 1.5 h and rising slowly to only 19% at 6 h. Insulin release of CS-NPs was similar to that of Tat-CS-NPs. In contrast, there is a burst release for PVA-NPs in simulated gastric fluid, reaching 40% at 0.25 h and rising fast to 80% at 6 h.
Figure 6. In vitro cumulative insulin release profiles of PVA-NPs, CS-NPs and Tat-CS-NPs in simulated gastric fluid (pH 1.2 hydrochloric acid solution) (A) and in simulated intestinal fluid (pH 7.4 PBS) (B), n = 3.
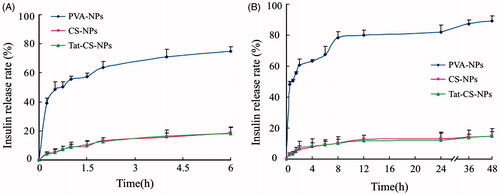
As shown in , in simulated intestinal fluid (pH 7.4 PBS), insulin release of CS-NPs was similar to that of Tat-CS-NPs which was very slow achieving only 15% at 48 h, while the drug release of PVA-NPs was drastic reaching 50% at 0.5 h and more than 80% at 48 h.
Regarding coumarin-6-loaded NPs (Tat-CS-NPs, CS-NPs, PVA-NPs), it was demonstrated that the cumulative release of coumarin-6 in HBSS was all below 5% at 6 h, with little leakage of coumarin-6.
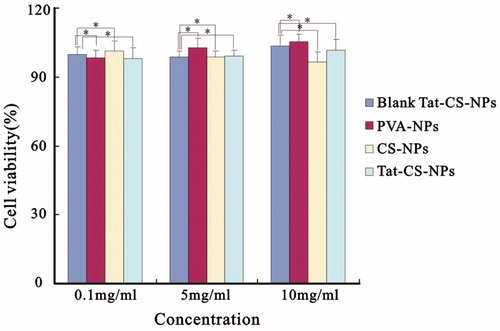
Cytotoxicity evaluations of NPs
The in vitro cytotoxicities of the four NPs are illustrated in . Cell viability at nanoparticle concentrations of 10 mg/ml (high concentration), 5 mg/ml (medium concentration) and 0.1 mg/ml (low concentration) was all greater than 90% and remained stable. These results suggested that Tat-CS-NPs at the concentration of 0.1–10 mg/ml were non-toxic.
Figure 7. In vitro cytotoxicity of Tat-CS-NPs, CS-NPs, PVA-NPs and Blank Tat-CS-NPs at nanoparticle concentrations ranging from 0.1 to 10 mg/ml, n = 3, *p < 0.05.
Uptake of NPs by Caco-2 cell monolayers
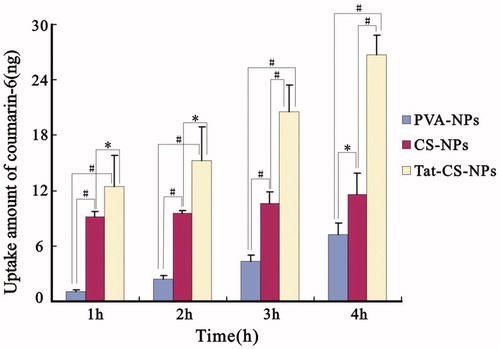
The fluorescent images in are shown that Tat-CS-NPs were internalized by Caco-2 cells more efficiently than CS-NPs and PVA-NPs. After the Caco-2 cell monolayer was incubated with coumarin-6-loaded NPs at 37 °C for 1 h, it was observed under fluorescent microscopy that Tat-CS-NPs () and CS-NPs () were of similar fluorescent intensities, both of which evidently exceeded that of PVA-NPs (). When the incubation time reached 2 h, the fluorescent intensity of Tat-CS-NPs () was remarkably higher than those of CS-NPs () and PVA-NPs (). According to , the uptake amount of coumarin-6 for Tat-CS-NPs, which was determined after Caco-2 cells were lysed and Tat-CS-NPs within were destroyed by acetone, was 12.44 ng at 1 h incubation time. In comparison, the uptake amount of coumarin-6 for PVA-NPs at 1 h incubation time was only 1.10 ng, which was much lower. As the incubation process went on, the uptake amounts of coumarin-6 for the three NPs were all increasing. At 2, 3 and 4 h incubation time, the uptake amounts of coumarin-6 were 15.20, 20.56 and 26.76 ng for Tat-CS-NPs, respectively, 9.51, 10.65 and 1.62 ng for CS-NPs, 2.45, 4.36 and 7.27 ng for PVA-NPs. It was calculated that the uptake amount of coumarin-6 for Tat-CS-NPs was about 2.3 times that for CS-NPs and 3.68 times that for PVA-NPs at 4 h incubation time. Accordingly, the uptake amount of Tat-CS-NPs was far higher than those of CS-NPs and PVA-NPs.
Figure 8. Fluorescence micrographs. Cellular uptake of coumarin-6-loaded PVA-NPs (A and D), coumarin-6-loaded CS-NPs (B and E) and coumarin-6-loaded Tat-CS-NPs (C and F) were observed under inverted fluorescence microscope after incubation with Caco-2 cell monolayers at 37 °C for 1 and 2 h, respectively. For PVA-NPs, green fluorescence at 2 h incubation time (D) resembles that at 1 h incubation time (A) under fluorescence microscope by visual observation; for CS-NPs, fluorescence at 2 h incubation time (E) is a bit stronger than that at 1 h incubation time (B); for Tat-CS-NPs, green fluorescence at 2 h incubation time (F) is markedly stronger than that at 1 h incubation time (C).
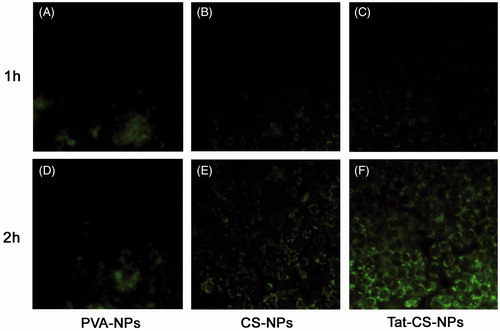
Figure 9. Uptake amount of coumarin-6. Uptake of coumarin-6-loaded NPs (PVA-NPs, CS-NPs and Tat-CS-NPs) by Caco-2 cells was evaluated at different incubation time (mean ± SD, n = 3) (*p < 0.05, #p < 0.01).
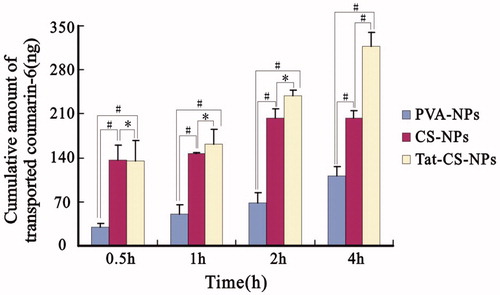
Transcellular transportation of NPs across Caco-2 cell monolayer
As indicated in , the cumulative amounts of transported coumarin-6 for Tat-CS-NPs at 0.5, 1 and 2 h incubation time were 134.34, 162.39 and 238.90 ng, and those for CS-NPs were 135.96, 146.35 and 202.86 ng. The two amounts at 0.5 h incubation time were similar. As the incubation process went on, the difference between the two amounts was increasing as can be seen at 1 and 2 h incubation time. When the Caco-2 cell monolayer was incubated with coumarin-6-loaded NPs at 37 °C for 4 h, there seemed to be much difference between the two groups. The former has drastically increased to 317.37 ng, while the latter only reached 203.58 ng. Besides, it can be seen from the chart that the cumulative amounts of transported coumarin-6 for Tat-CS-NPs and CS-NPs at 0.5, 1, 2 and 4 h were a lot higher than those of PVA-NPs at the same time points which were 29.35, 49.73, 68.88 and 111.73 ng, respectively.
Figure 10. Cumulative amount of transported coumarin-6. Transportation performance of coumarin-6-loaded NPs (PVA-NPs, CS-NPs and Tat-CS-NPs) across the Caco-2 cell monolayer were evaluated at different incubation time (mean ± SD, n = 3) (*p < 0.05, #p < 0.01).
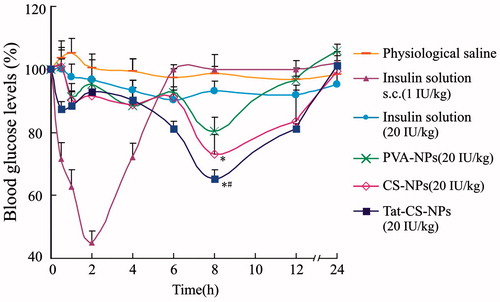
Pharmacodynamic studies
The hypoglycemic effect of Tat-CS-NPs was evaluated on diabetic rats in comparison with those of subcutaneous injection of insulin solution, colon delivery of physiological saline, insulin solution, PVA-NPs and CS-NPs. As can be seen from , as for colon delivery of PVA-NPs, CS-NPs and Tat-CS-NPs (20 IU/kg), the most significant hypoglycemic effects were observed at 8 h and the blood glucose levels were 80.42%, 73.01% and 65.37%, respectively, which means Tat-CS-NPs produced the most remarkable hypoglycemic effect. It was calculated that the D% of Tat-CS-NPs was 1.79-fold higher than that of CS-NPs, 6.89-fold higher than that of PVA-NPs. ANOVA was used for statistical analysis. The results showed that the hypoglycemic effect of Tat-CS-NPs was significantly different from that of CS-NPs (p < 0.05). When compared with PVA-NPs, the hypoglycemic effect of Tat-CS-NPs was more significantly different (p < 0.01). Moreover, unlike subcutaneous injection of insulin solution, Tat-CS-NPs could maintain the effect for a longer period.
Figure 11. Blood glucose levels of diabetic rats. Blood glucose levels of diabetic rats were measured at particular intervals following subcutaneous injection (s.c.) of insulin solution at the dose of 1 IU/kg, colon delivery of physiological saline, and colon delivery of insulin solution, PVA-NPs, CS-NPs and Tat-CS-NPs at the dose of 20 IU/kg, each data point represents the mean ± SD (n = 3) (*p < 0.05, #p < 0.01).
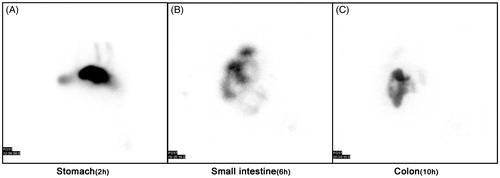
Gamma-scintigraphy
displays the visualization of the running behaviors of 99mTc isotope labeled Tat-CS-NPs in mini-pigs' digestive tract at 2, 6 and 10 h, respectively, after oral delivery of the drug. shows clearly that 99mTc labeled Tat-CS-NPs reached the pig's stomach at about 2 h. demonstrates that the drug was in the pig's small intestine at approximately 6 h. reveals that the nanoparticles were located at colon lumen in the pig at 10 h, which suggests Tat-CS-NPs could successfully arrive at colon.
Figure 12. Scintigraphic imaging. SPECT scan on mini-pigs' stomach, small intestine and colon at 2 h (A), 6 h (B) and 10 h (C), respectively, after oral delivery of 99mTc-labeled Tat-CS-NPs (n = 3).
Discussion
It is of wide acceptance that colon is suited for protein and peptide drug absorption. Compared with stomach and small intestine, colon is boasted of low intensity and activity of digestive enzymes and neutral pH value, which enable proteins and peptides not to be hydrolyzed and degraded (Varum et al., Citation2008). Besides, the apical cell membrane in colon exhibits a greater degree of fluidity in comparison with that in small intestine, accounting for its high responsiveness to permeation enhancers (Masaoka et al., Citation2006; Philip & Philip, Citation2010). Moreover, colon may be more appropriate to be developed into a bioadhesive system than stomach and small intestine are, due to a thicker mucus layer and more moderate colonic motility (Varum et al., Citation2008).
However, it is undeniable that some barriers against the drug diffusion and permeation exist in colon, which exert a negative influence on colonic absorption of the drug (Philip & Philip, Citation2010; Maroni et al., Citation2012). Many attempts have been conducted to overcome the obstacles. For example, absorption enhancers were used to promote the drug permeation and absorption, with definite toxicity and with little effect on overcoming the barrier against the drug diffusion (Patel, Citation2013). Due to the fact that the mucous layer in colon is thick and turnover rate slowly and it is insensitive to mucus secretion stimulation (Maroni et al., Citation2012), the benefits of mucoadhesive polymeric systems were exploited, while the barrier against the drug penetration remained to be overcome (Van den Mooter, Citation2006; McGirr et al., Citation2009).
The present study used Tat-CS-NPs as carriers to boost colonic absorption of oral protein and peptide drugs. On the one hand, Tat-modified nanoparticles as colon-specific delivery system were supposed to overcome the barrier against the drug penetration thereby increase the drug absorption; on the other hand, the modification of a-CS was supposed to give the nanoparticles good bioadhesive ability conquering the barrier against the drug diffusion. Another point is that a-CS was supposed to protect Tat from being degraded in stomach and small intestine, and helped maintain its activity for transcellular transportation of the nanoparticles in colonic epithelial cells.
As indicated by a number of researches, chitosan and it synthetic derivatives were endowed with good bioadhesive ability, and they were applied in colonic bioadhesive system and colon-specific drug delivery system (Kotzé et al., Citation1997; Van der Merwe et al., Citation2004; Bonferoni et al., Citation2009). In this study, N,N,N-trimethyl-N-dodecyl chitosan was synthesized with chitosan as the raw material, in consideration that the introduction of N-trimethyl might increase its water-solubility and permeability in neutral pH environment (Chen et al., Citation2008; Verheul et al., Citation2008; Yin et al., Citation2009), and the introduction of dodecyl might improve its emulsifying capacity. Besides, N,N,N-trimethyl-N-dodecyl chitosan might potentiate the steric hindrance effect of chitosan thus effectively protect Tat against degradation in GIT. It was demonstrated yet unpublished in our research group that a protective effect of less than 20% degradation of Tat during 24 h was obtained in simulated intestinal fluid containing trypsin (pH 6.8), and the same protective effect was acquired during 48 h in simulated intestinal fluid containing colonic contents (pH 7.4).
This study for the first time used N,N,N-trimethyl-N-dodecyl chitosan as emulsifier for the preparation of NPs with PLGA as matrix material. Currently, preparation of PLGA nanoparticles modified with chitosan and its synthetic derivatives has been reported, in which chitosan and its derivatives were adhered to the surface of PLGA nanoparticles through weak interaction such as static electricity and Van der Waals force, and they were apt to dissociate owing to the harsh environment in GIT (Iyer et al., Citation2010; Chen et al., Citation2011). Here, using a-CS as emulsifier, the alkyl chain of a-CS might embed in matrix of PLGA nanoparticles, making a-CS on the surface of nanoparticles not easy to detach. Besides, a-CS possessed strong surfactivity which could reduce the surface energy of the nanoparticles and raise the stability of them.
Particle size and morphology observation indicated that Tat-CS-NPs were of roughly the same size below 160 nm and stable which enabled them to easily cross physiological barriers. As chitosan and Tat were positively charged, the nanoparticles possessed a zeta potential above 50 mV, making them apt to bind to negatively charged epithelial cells in GIT thus promote drug permeation. Meanwhile, PVA-NPs had an almost neutral zeta potential as PVA were electrically neutral.
As the in vitro release study demonstrated that, in simulated gastric fluid and simulated intestinal fluid, a cumulative release of less than 20% was displayed at 6 h as was displayed at 48 h for both Tat-CS-NPs and CS-NPs, while a burst release above 40% emerged for PVA-NPs at 0.5 h. That is probably because most insulin was encapsulated in the former, while a large amount of insulin adhered to the surface of the latter. Consequently, insulin in Tat-CS-NPs and CS-NPs released little, while that adherent to the surface of PVA-NPs released rapidly. Another possible reason is that a-CS modification and PLGA as matrix give Tat-CS-NPs and CS-NPs good stability and enabled them not to be dissolutioned in the environment thus protect biodrugs within from exposure and release. It was supposed in this study that insulin-loaded nanoparticles be absorbed in colon, and the above experiment fully demonstrated that this expectation could be met.
As human colon carcinoma cells, Caco-2 monolayers are generally used as a model for the small intestinal mucosa. Since their structure resembles that of colonic epithelial cells, they were harnessed for the evaluation of the colon absorption of oral protein and peptide drugs in this study. The Caco-2 cell experiment verified that transcellular transportation capacity and cellular uptake amount of Tat-CS-NPs were even better than those of PVA-NPs and CS-NPs. The reason may be that Tat helped the nanoparticles penetrate into Caco-2 cells and pass through Caco-2 cell monolayers, and a-CS opened the tight junctions thereby enabled entrance of the nanoparticles through paracellular pathway (Artursson et al., Citation1994).
At present, many studies demonstrated that bioadhesive systems in stomach and small intestine were developed with the aid of chitosan and its derivatives. Despite promising results were obtained in small animals, primarily rats, the results obtained in humans have not fulfilled expectations (Kararli, Citation1995; Varum et al., Citation2008). That is possibly owing to the differences between human beings' digestive tract and small animals' in terms of anatomical structure, physiological characteristics, gastrointestinal motility, mucus turnover rate, gastric emptying rate and transit time (Kararli, Citation1995; Varum et al., Citation2008). For humans, gastrointestinal motility is more violent and mucus turnover rate is quicker, as compared with small animals (Kararli, Citation1995; Varum et al., Citation2008).
In order to more exactly study hypoglycemic effect of Tat-CS-NPs loaded with insulin, the drug was directly ingested into the ascending colon of diabetic rats after surgical procedure with small incision in their abdomen cavity portion (Bayat et al., Citation2008), avoiding the occurrence that the nanoparticles adhere to small intestinal mucosa of the rats and fail to run into colon following oral administration. The experimental results indicated that the hypoglycemic effect of CS-NPs was much better than that of PVA-NPs. The reason for that, on the one hand, probably was a-CS on the surface of the nanoparticles, provided with favorable bioadhesive capability, made the nanoparticles adhere to colonic mucosa lengthening the drug retention time. The bioadhesive ability of the nanoparticles has been determined through isolated rat colon test and turbidimetric analysis in our research group, and the results indicated that Tat-CS-NPs possessed good colonic bioadhesive ability (data not shown). On the other hand, a-CS might open the tight junctions between epithelial cells enabling entrance of the nanoparticles in paracellular pathway, hence boosted drug absorption (Gulbake & Jain, Citation2012; Zhang et al., Citation2012). In addition, the results from drug release in vitro showed that insulin released from PVA-NPs was much faster than those from Tat-CS-NPs and CS-NPs. More than 40% release in less than 30 min at pH7.4 media was observed from PVA-NPs. Free insulin released from PVA-NPs might not be effectively absorbed due to its inherent high molecular weight and hydrophilicity as well as poor biomembrane permeability. Furthermore, the hypoglycemic effect of Tat-CS-NPs was proved better than that of CS-NPs. That is probably due to the improved transcellular transportation capacity and penetration ability in colonic epithelial cells given by CPPs (Tat) (Hällbrink et al., Citation2001; Torchilin, Citation2008). It has been reported in some researches that CPPs modified nanoparticles could facilitate insulin penetration in small intestinal epithelial cells and promote insulin absorption in small intestine (Kamei et al., Citation2008; Liu et al., Citation2013). Compared with unmodified nanoparticles, more pronounced hypoglycemic effects were observed (with blood glucose level lowered by 2.5-times for l-R8 and 3.7-times for d-R8) (Liu et al., Citation2013). The experimental results shown the D% of Tat-CS-NPs were 1.79-fold higher than that of CS-NPs, 6.89-fold higher than that of PVA-NPs. It was concluded from the above study that Tat-CS-NPs had the best hypoglycemic effect of all the three nanoparticles, which fully demonstrated that nanoparticles with the modification of Tat and chitosan together could boost colonic absorption of oral protein and peptide drugs. An interesting phenomenon was observed based on the results. For no matter PVA-NPs, CS-NPs or Tat-CS-NPs, a remarkable hypoglycemic effect appeared simultaneously at 8 h. A possible cause was that colon moves slowly and has a thick mucus layer, so that the nanoparticles could pass through the mucous layer and enter into epithelial cells rather slowly, and they might be released and produce pharmacological effect a certain time after passing through the colonic epithelial cells and entering blood circulation.
In order to confirm Tat-CS-NPs could reach colon, mini-pigs, whose GIT is similar to human being's in terms of anatomical, physiological and biochemical characteristics, gastrointestinal motility and mucus turnover rate, were chosen as animal models. SPECT scan was utilized to visualize how the 99mTC isotope labeled Tat-CS-NPs traveled in mini-pigs' GIT at particular intervals after oral administration (Ashford & Fell, Citation1994; Soane et al., Citation1999; Ergün et al., Citation2000; Polyák et al., Citation2013). Based on oral administration time, drug residence time in GIT (generally in stomach 2 h after oral administration, in small intestine 6 h after oral administration, in colon 10 h after oral administration), as well as the scintigraphic images, it could be inferred that Tat-CS-NPs arrived at pigs' colon.
Conclusion
This study for the first time indicated that modified nanoparticles (Tat-CS-NPs) with CPPs (Tat) and amphipathic chitosan derivative as carriers could enhance colonic absorption of oral protein and peptide drug. Cellular uptake and transcellular transportation performance of Tat-CS-NPs were proved better than those of CS-NPs, and even better than those of PVA-NPs. In addition, it was demonstrated that Tat-CS-NPs loaded with insulin produced the most significant hypoglycemic effect after being ingested into rats' colon, suggesting improved penetration efficiency given by Tat. Gama scintigraphy further proved that Tat-CS-NPs could arrive at colon. Hence, it is believed that Tat-CS-NPs as carriers may provide an effective means for oral colon delivery of protein and peptide drugs, overcoming the barriers against the drug diffusion and penetration in colon. The findings may spark a new line of thinking on oral protein and peptide drug development.
Acknowledgement
We acknowledged Qing Zhang (The First Affiliated Hospital of Nanchang University) for his kind technical support of gamma-scintigraphy using SPECT.
Declaration of interest
The authors report no conflicts of interest. This work was supported by the National Natural Science Foundation of China (Grant Nos. 81260482 and 30960466) and the Natural Science Foundation of Jiangxi Province, China (Grant No. 20122BAB205031).
References
- Arbit E. (2004). The physiological rationale for oral insulin administration. Diabetes Technol Ther 6:510–17
- Artursson P, Lindmark T, Davis SS, Illum L. (1994). Effect of chitosan on the permeability of monolayers of intestinal epithelial cells (Caco-2). Pharm Res 11:1358–61
- Ashford M, Fell JT. (1994). Targeting drugs to the colon: delivery systems for oral administration. J Drug Target 2:241–58
- Bakhru SH, Furtado S, Morello AP, Mathiowitz E. (2013). Oral delivery of proteins by biodegradable nanoparticles. Adv Drug Deliv Rev 65:811–21
- Bayat A, Dorkoosh FA, Dehpour AR, et al. (2008). Nanoparticles of quaternized chitosan derivatives as a carrier for colon delivery of insulin: ex vivo and in vivo studies. Int J Pharm 356:259–66
- Bernkop-Schnürch A, Dünnhaupt S. (2012). Chitosan-based drug delivery systems. Eur J Pharm Biopharm 81:463–9
- Bonferoni MC, Sandri G, Rossi S, et al. (2009). Chitosan and its salts for mucosal and transmucosal delivery. Expert Opin Drug Deliv 6:923–39
- Chauhan A, Tikoo A, Kapur AK, Singh M. (2007). The taming of the cell penetrating domain of the HIV Tat: myths and realities. J Control Release 117:148–62
- Chen MC, Sonaje K, Chen KJ, Sung HW. (2011). A review of the prospects for polymeric nanoparticle platforms in oral insulin delivery. Biomaterials 32:9826–38
- Chen F, Zhang ZR, Yuan F, et al. (2008). In vitro and in vivo study of N-trimethyl chitosan nanoparticles for oral protein delivery. Int J Pharm 349:226–33
- Cui FD, Shi K, Zhang LQ, et al. (2006). Biodegradable nanoparticles loaded with insulin–phospholipid complex for oral delivery: preparation, in vitro characterization and in vivo evaluation. J Control Release 114:242–50
- Damgé C, Maincent P, Ubrich N. (2007). Oral delivery of insulin associated to polymeric nanoparticles in diabetic rats. J Control Release 117:163–70
- Ensign LM, Cone R, Hanes J. (2012). Oral drug delivery with polymeric nanoparticles: the gastrointestinal mucus barriers. Adv Drug Deliv Rev 64:557–70
- Ergün EL, Ercan MT, Selek H, et al. (2000). Evaluation of 99mTc labelled poly lactic acid microspheres for diagnostic radioembolization. J Microencapsul 17:509–18
- Foged C, Nielsen HM. (2008). Cell-penetrating peptides for drug delivery across membrane barriers. Expert Opin Drug Deliv 5:105–17
- Gulbake A, Jain SK. (2012). Chitosan: a potential polymer for colon-specific drug delivery system. Expert Opin Drug Deliv 9:713–29
- Hällbrink M, Florén A, Elmquist A, et al. (2001). Cargo delivery kinetics of cell-penetrating peptides. Biochim Biophys Acta 1515:101–9
- Haupt S, Rubinstein A. (2002). The colon as a possible target for orally administered peptide and protein drugs. Crit Rev Ther Drug Carrier Syst 19:499–551
- He B, Lin P, Jia ZR, et al. (2013). The transport mechanisms of polymer nanoparticles in Caco-2 epithelial cells. Biomaterials 34:6082–98
- Hejazi R, Amiji M. (2003). Chitosan-based gastrointestinal delivery systems. J Control Release 89:151–65
- Iyer H, Khedkar A, Verma M. (2010). Oral insulin – a review of current status. Diabetes Obes Metab 12:179–85
- Jin Y, Song YP, Zhu X, et al. (2012). Goblet cell-targeting nanoparticles for oral insulin delivery and the influence of mucus on insulin transport. Biomaterials 33:1573–82
- Johnson RM, Harrison SD, Maclean D. (2011). Therapeutic applications of cell-penetrating peptides. Methods Mol Biol 683:535–51
- Kamei N, Morishita M, Eda Y, et al. (2008). Usefulness of cell-penetrating peptides to improve intestinal insulin absorption. J Control Release 132:21–5
- Kamei N, Onuki Y, Takayama K, Takeda-Morishita M. (2013). Mechanistic study of the uptake/permeation of cell-penetrating peptides across a Caco-2 monolayer and their stimulatory effect on epithelial insulin transport. J Pharm Sci 102:3998–4008
- Kaplan IM, Wadia JS, Dowdy SF. (2005). Cationic TAT peptide transduction domain enters cells by macropinocytosis. J Control Release 102:247–53
- Kararli TT. (1995). Comparison of the gastrointestinal anatomy physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm Drug Dispos 16:351–80
- Khafagy El-S, Morishita M. (2012). Oral biodrug delivery using cell-penetrating peptide. Adv Drug Deliv Rev 64:531–9
- Khafagy El-S, Morishita M, Onuki Y, Takayama K. (2007). Current challenges in non-invasive insulin delivery systems: a comparative review. Adv Drug Deliv Rev 59:1521–46
- Koren E, Torchilin VP. (2012). Cell-penetrating peptides: breaking through to the other side. Trends Mol Med 18:385–92
- Kotzé AF, Lueβen HL, De Leeuw BJ, et al. (1997). N-Trimethyl chitosan chloride as a potential absorption enhancer across mucosal surfaces: in vitro evaluation in intestinal epithelial cells (Caco-2). Pharm Res 14:1197–202
- Liu XL, Liu C, Zhang WJ, et al. (2013). Oligoarginine-modified biodegradable nanoparticles improve the intestinal absorption of insulin. Int J Pharm 448:159–67
- Maroni A, Zema L, Del Curto MD, et al. (2012). Oral colon delivery of insulin with the aid of functional adjuvants. Adv Drug Deliv Rev 64:540–56
- Masaoka Y, Tanaka Y, Kataoka M, et al. (2006). Site of drug absorption after oral administration: assessment of membrane permeability and luminal concentration of drugs in each segment of gastrointestinal tract. Eur J Pharm Sci 29:240–50
- McGirr ME, McAllister SM, Peters EE, et al. (2009). The use of the InteliSite® companion device to deliver mucoadhesive polymers to the dog colon. Eur J Pharm Sci 36:386–91
- Milletti F. (2012). Cell-penetrating peptides: classes, origin, and current landscape. Drug Discov Today 17:850–60
- Patel MM. (2011). Cutting-edge technologies in colon-targeted drug delivery systems. Expert Opin Drug Deliv 8:1247–58
- Patel MM. (2013). Colon targeting: an emerging frontier for oral insulin delivery. Expert Opin Drug Deliv 10:731–9
- Peppas NA, Kavimandan NJ. (2006). Nanoscale analysis of protein and peptide absorption: insulin absorption using complexation and pH-sensitive hydrogels as delivery vehicles. Eur J Pharm Sci 29:183–97
- Philip AK, Philip B. (2010). Colon targeted drug delivery systems: a review on primary and novel approaches. Oman Med J 25:70–8
- Polyák A, Hajdu I, Bodnár M, et al. (2013). 99mTc-labelled nanosystem as tumour imaging agent for SPECT and SPECT/CT modalities. Int J Pharm 449:10–17
- Reissmann S. (2014). Cell penetration: scope and limitations by the application of cell-penetrating peptides. J Pept Sci 20:760–84
- Rekha MR, Sharma CP. (2013). Oral delivery of therapeutic protein/peptide for diabetes – future perspectives. Int J Pharm 440:48–62
- Santra S, Yang H, Stanley JT, et al. (2005). Rapid and effective labeling of brain tissue using TAT-conjugated CdS: Mn/ZnS quantum dots. Chem Commun 25:3144–6
- Shelma R, Sharma CP. (2011). Submicroparticles composed of amphiphilic chitosan derivative for oral insulin and curcumin release applications. Colloids Surf B Biointerfaces 88:722–8
- Shi CM, Zhu Y, Ran XZ, et al. (2006). Therapeutic potential of chitosan and its derivatives in regenerative medicine. J Surg Res 133:185–92
- Sinha VR, Singh A, Kumar RV, et al. (2007). Oral colon-specific drug delivery of protein and peptide drugs. Crit Rev Ther Drug Carrier Syst 24:63–92
- Skyler JS. (2007). Prediction and prevention of type 1 diabetes: progress, problems and prospects. Clin Pharmacol Ther 81:768–71
- Soane RJ, Frier M, Perkins AC, et al. (1999). Evaluation of the clearance characteristics of bioadhesive systems in humans. Int J Pharm 178:55–65
- Subramanian S, Pandey U, Gugulothu D, et al. (2013). Modification of PLGA nanoparticles for improved properties as a 99mTc-labeled agent in sentinel lymph node detection. Cancer Biother Radiopharm 28:598–606
- Torchilin VP. (2008). Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv Drug Deliv Rev 60:548–58
- Trehan A, Ali A. (1998). Recent approaches in insulin delivery. Drug Dev Ind Pharm 24:589–97
- Tréhin R, Krauss U, Beck-Sickinger AG, et al. (2004). Cellular uptake but low permeation of human calcitonin-derived cell penetrating peptides and Tat (47–57) through well-differentiated epithelial models. Pharm Res 21:1248–56
- Van den Mooter GV. (2006). Colon drug delivery. Expert Opin Drug Deliv 3:111–25
- Van der Merwe SM, Verhoef JC, Verheijden JHM, et al. (2004). Trimethylated chitosan as polymeric absorption enhancer for improved peroral delivery of peptide drugs. Eur J Pharm Biopharm 58:225–35
- Varum FJO, McConnell EL, Sousa JJS, et al. (2008). Mucoadhesion and the gastrointestinal tract. Crit Rev Ther Drug Carrier Syst 25:207–58
- Verheul RJ, Amidi M, Van der Wal S, et al. (2008). Synthesis, characterization and in vitro biological properties of O-methyl free N,N,N-trimethylated chitosan. Biomaterials 29:3642–9
- Wang FH, Wang Y, Zhang X, et al. (2014). Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. J Control Release 174:126–36
- Wild S, Roglic G, Green A, et al. (2004). Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 27:1047–53
- Yang LB, Chu JS, Fix JA. (2002). Colon-specific drug delivery: new approaches and in vitro/in vivo evaluation. Int J Pharm 235:1–15
- Yin LC, Ding JY, He CB, et al. (2009). Drug permeability and mucoadhesion properties of thiolated trimethyl chitosan nanoparticles in oral insulin delivery. Biomaterials 30:5691–700
- Zhang H, Alsarra IA, Neau SH. (2002). An in vitro evaluation of a chitosan-containing multiparticulate system for macromolecule delivery to the colon. Int J Pharm 239:197–205
- Zhang C, Ding Y, Ping QN, Yu LL. (2006). Novel chitosan-derived nanomaterials and their micelle-forming properties. J Agric Food Chem 54:8409–16
- Zhang XY, Sun MZ, Zheng AP, et al. (2012). Preparation and characterization of insulin-loaded bioadhesive PLGA nanoparticles for oral administration. Eur J Pharm Sci 45:632–8
- Zhang P, Zhang XZ, Brown J, et al. (2010). Global healthcare expenditure on diabetes for 2010 and 2030. Diabetes Res Clin Pract 87:293–301