
Abstract
Context: Muscle spasm needs prompt relief of symptoms. Chlorzoxazone is a centrally muscle relaxant.
Objectives: The aim of this study was to prepare chlorzoxazone orodispersible tablets (ODTs) allowing the drug to directly enter the systemic circulation and bypassing the first-pass metabolism for both enhancing its bioavailability and exerting a rapid relief of muscular spasm.
Materials and methods: ODTs were prepared by direct compression method using Pharmaburst®500, Starlac®, Pearlitol flash®, Prosolv® odt and F-melt® as co-processed excipients. Three ratios of the drug to the other excipients were used (0.5:1, 1:1 and 2:1).
Results and Discussion: All ODTs were within the pharmacopeial limits for weight and content. ODTs containing Pharmaburst®500 showed the shortest wetting time (∼45.33 s), disintegration time (DT) (∼43.33 s) and dissolution (Q15min 100.63%). By increasing the ratio of CLZ: Pharmaburst®500 from 0.5:1 to 1:1 and 2:1, the DT increased from 26.43 to 28.0 and 43.33 s, respectively. By using Prosolv® odt, ODTs failed to disintegrate in an acceptable time >180 s. DT of ODTs using different co-processed excipients can be arranged as follows: Pharmaburst® 500 < F-melt® <Pearlitol flash® <Starlac® <Prosolv® odt. Pharmacokinetic study of the optimum formula F1 (50 mg CLZ) in rabbits using HPLC-UV detector revealed a shorter Tmax (0.333 h) compared with Myofen® capsules (250 mg CLZ) (1.083 h) which is considered a promising treatment, especially for the rapid relief of muscle spasm.
Conclusion: It could be concluded that orodispersible tablets are a promising carrier for CLZ designed for management of muscle spasm due to the enhanced dissolution and rapid absorption of the drug through the oral mucosa.
Introduction
Muscle spasm is a painful reflex contraction of muscle that can cause involuntary movement and interfere with muscle function, causing defacement. It can be caused by musculoskeletal conditions or by neurological conditions associated with upper motor neuron lesions (spasticity) (Sweetman et al., Citation2009). Common skeletal muscle relaxants have been approved for either treatment of spasticity or for treatment of musculoskeletal conditions. Only baclofen, dantrolene and tizanidine are approved by FDA for the treatment of spasticity, but carisoprodol, chlorzoxazone, cyclobenzaprine, metaxalone, methocarbamol and orphenadrine have been approved for treatment of musculoskeletal disorders (Chou et al., Citation2004).
Chlorzoxazone (CLZ) is a centrally acting musculoskeletal relaxant with sedative properties. Its chemical name is 5-chloro-2(3H)-benzoxazolone, with low solubility in water. After oral administration, CLZ is completely absorbed and is rapidly metabolized in the liver to 6-hydroxychlorzoxazone which has little or no muscle relaxant activity when tested in mice and rats (Florey et al., Citation1987). CLZ inhibits muscle spasm by exerting an effect primarily at the level of the spinal cord and subcortical areas of the brain. Its effect begins within an hour after an oral dose and lasts for 3–4 h. The usual initial oral dose is 500 mg three or four times daily; though the dose can often be reduced subsequently to 250 mg three or four times daily. CLZ is usually given with analgesics in compound preparations. The most common side effects of CLZ are drowsiness, dizziness and headache (Sweetman et al., Citation2009). CLZ belongs to biopharmaceutics classification (BCS) class II, where its solubility is poor in gastrointestinal fluid and showing good permeability. Low solubility and dissolution rate of poorly water-soluble drugs make them suffer from limited clinical effect (Xu et al., Citation2014).
In 1970s, fast-dissolving dosage forms (FDDFs) were developed as an alternative to oral solid dosage forms for pediatric and geriatric patients who have difficulties in swallowing solid dosage forms (Seager, Citation1998; Nagaraju et al., Citation2013). FDDFs have unique properties and advantages like rapid disintegration and dissolution in the oral cavity without the need for water, accurate dosing, rapid onset of action, pleasant taste and improved patient compliance, especially for pediatrics and geriatrics (Heer et al., Citation2013). In addition, they show advantage of avoidance of first-pass hepatic metabolism by pre-gastric absorption, which improves bioavailability and safety profiles of drugs that have significant amounts of toxic metabolites mediated by first-pass liver metabolism and gastric metabolism (Wilson et al., Citation1987). But FDDFs are not suitable for drugs with relatively larger doses >400 mg, and are not suitable for patients with Jorgen’s syndrome (a disorder of immune system identified by its two most common symptoms – dry eyes and a dry mouth) or under concurrent anticholinergic medications (Chang et al., Citation2000). There are many forms of FDDFs such as: orodispersible tablets (ODTs) (Heer et al., Citation2013), wafers (Alli, Citation2011) and oral dissolving films (Honey et al., Citation2008; Dixit & Puthli, Citation2009). There are several methods for preparation of ODTs, such as direct compression, molding, spray drying, sublimation and lyophilization (Bolhuis et al.,Citation1997; Sammour et al., Citation2006; Patel et al., Citation2007; Suresh et al., Citation2007). Different strategies were used to induce fast disintegration of ODTs, and one of them is the incorporation of disintegrants (Al-Khattawi & Mohammed, Citation2013). Many ODTs are highly porous, and if the processing parameters are not optimized, the tablets can exhibit higher friability, irrespective of any hardness changes. Thus, in order to produce mechanically hard ODTs without compromising the disintegration time, co-processed excipients must be used.
CLZ is commercially available in the Egyptian market in dosage forms as conventional tablets (Myoflex®) or capsules (Myofen®), but it is not available as ODTs. In this study, our goal was to develop ODTs of CLZ by using different co-processed excipients to provide rapid drug release and consequently fast relief of muscle spasm and to assess the bioavailability of the best chosen ODT in rabbits.
Experimental
Materials
Chlorzoxazone powder was received as a kind gift from EVA pharmaceutical industries (Cairo, Egypt). Pharmaburst®500 and Lubripharm® Ssf (Sodium stearyl fumarate) were a kind gift from SPI pharma (Wilmington, DE). Starlac® and Peralitol flash® were obtained from Roquette (Lestrem, France). Prosolv® odt was a kind gift from JRS pharma GmbH& Co. KG (Rosenberg, Germany). F-melt® was a kind gift from Fuji Chemical Industry Ltd. (Toyama-Pref, Japan. Mono ammonium glycyrrhizinate (MAG) was obtained from Indena S.P.A (Milan, Italy). Acetonitrile and methanol (HPLC grade) were from Sigma Aldrich (St. Louis, MO). All other chemicals were of analytical grade.
Methods
Preparation of orodispersible tablets of chlorzoxazone
Fifteen ODT formulations of CLZ were prepared by direct compression method as shown in . The drug and the directly compressible excipients were mixed by adding small portion of each at a time and blended to get a uniform mixture. Three ratios of the drug to the other excipients were used (0.5:1, 1:1 and 2:1). The powder blend was then lubricated with Lubripharm®, sweetened with MAG and compressed into tablets of 150 mg using 7 mm curved face by a single punch tablet machine (Royal Artist, Mumbai, India).
Table 1. Composition of different ODTs prepared by direct compression using different co-processed excipients.
Evaluation of the prepared tablets
Weight variation
Twenty tablets were selected randomly from each formulation and weighed individually. The individual weights were compared with the mean weight and the standard deviation (SD) was calculated.
Thickness variation
Ten tablets from each formulation were taken randomly and their thickness was measured using micrometer (Starrett, Athol, MA), then the mean thickness and SD were calculated.
Friability test
Five tablets of each formula were weighed and placed in a Friabilator (Mumbai, India) at 25 rpm for 4 min. The tablets were reweighed and the percent friability was then calculated according to the following equation:
(1)
Hardness test
The hardness of three tablets from each formulation was tested using Monsanto hardness tester (Creve Coeur, MO) and the mean and SD values were calculated.
Drug content
A tablet containing drug was dissolved in simulated saliva fluid (SSF, pH 6.8) [sodium chloride (0.8% w/v), sodium phosphate dibasic (0.238% w/v) and potassium phosphate monobasic (0.019% w/v)] and analyzed spectrophotometrically at 281 nm. The concentration of CLZ in the tablet was deduced from a pre-constructed calibration curve (R2 = 0.998, n = 3). Drug content was determined in triplicate for each formulation batch and the results were expressed as mean ± SD.
Wetting time
A piece of tissue paper folded twice was placed in a small petri dish containing 6 mL of dye solution (methylene blue aqueous solution). A tablet was carefully placed on the surface of the paper and the time required for the dye solution to reach the upper surface of the tablet was noted as the wetting time (WT). Any value more than 3 min was considered a slow WT. The WT for each formulation was carried out in triplicate and the results were expressed as mean ± SD.
In vitro disintegration time
One tablet was placed in a beaker containing 5 mL SSF (pH 6.8) at 37 ± 0.5 °C and the time required for complete disintegration of tablet was determined. In vitro disintegration time (DT) for each formulation was carried out in triplicate and the results were expressed as mean ± SD.
In vitro dissolution
In vitro dissolution studies were performed using Type II (paddle) dissolution apparatus at 50 rpm and 900 mL of SSF (pH 6.8) as a dissolution medium. Temperature of dissolution medium was maintained at 37 ± 0.5 °C. Aliquots of the dissolution medium (5 mL) were withdrawn at specific time intervals (2, 4, 6, 10, 15, 20, 25, 30, 45 and 60 min) and were replaced immediately with equal volume of fresh medium kept at the same temperature. The samples were filtered through 0.22 mm membrane filters, diluted suitability and analyzed spectrophotometrically at 281 nm. Drug concentration was expressed as cumulative percent drug dissolved. In vitro dissolution for each formulation was carried out in triplicate and the results were expressed as mean ± SD.
Differential scanning calorimetry
Differential scanning calorimetry (DSC) was performed with differential scanning calorimeter (model TA-50 WSI, Shimadzu, Japan) calibrated with indium. Samples were placed in flat-bottomed aluminum pan and heated at constant rate of 10 °C/min in an inert nitrogen atmosphere at temperature range of 10–300 °C. The DSC thermograms were performed for pure CLZ, Pharmaburst®500, Lubripharm®, MAG and on the selected ODTs.
In vivo bioavailability study
Study design
Twelve male white New Zealand rabbits (weighing 2–2.5 kg) were used in the study and were randomly divided into two groups of equal size using parallel design. The experiment was performed in accordance with ethical procedures and policies approved by the Animal Care and Use Committee of the Faculty of Pharmacy, Cairo University, Cairo, Egypt, (PI = 1117) following the 18th World Medical Association General Assembly, Helsinki, June 1964, and updated by the 59th World Medical Association General Assembly, Seoul, October 2008. The study was carried out to compare the pharmacokinetics of CLZ from selected ODTs (F1) containing 50 mg CLZ to a commercially available Myofen® capsules (250 mg CLZ, EVA PHARMA). Food was withdrawn 10 h prior the in vivo study with water ad libitum, and no food was allowed 4 h after dosing. Rabbits of group A were orally administered with Myofen® capsules, whereas those of group B were administered with our prepared ODT (F1). For rabbits of group B, the ODT was placed between the jaws of tweezers and inserted onto the oral cavity and was held by the tweezers until it dissolved. A small incision in the marginal ear vein was made and blood samples (2 mL) were collected into heparinized tubes at the following time points: 0 (pre-dose), 0.25, 0.5, 1, 1.5, 2, 3, 4, 6 and 8 h after administration of the treatment. Plasma was obtained by centrifugation of blood samples at 4000 rpm for 10 min and kept frozen at −20 °C until analysis.
HPLC analysis
(a) Chromatographic conditions: Plasma concentrations of CLZ were determined using an HPLC method (Zhang & Xu, Citation2003) with slight modification. The column was XTerra® PR18, 5 μm, 4.6 mm × 250 mm (Waters Corp., Ireland). The mobile phase consisted of methanol and 0.05M dihydrogen phosphate (50:50) delivered at a flow-rate of 1.5 mL/min. The effluent was monitored at 280 nm using a SPD-10A VP (Shimadzu, Japan) UV visible detector. Sample preparation and analysis were performed at ambient temperature.
(b) Preparation of plasma samples: Plasma (0.3 mL) with 0.3 mL acetonitrile and 0.3 mL methanol were vortex-mixed for 30 s. The samples were centrifuged at 4000 rpm for 5 min. The upper layer was then transferred to another tube, and 20 μl were injected into the column for analysis.
Pharmacokinetic analysis
Pharmacokinetic characteristics from plasma data following administration of the two treatments were estimated for all rabbits by using a computer program, WinNonlin version 1.5 (Scientific consulting Inc., Cary, NC). Non-compartmental analysis was used. A drug plasma level versus time curve was constructed. The estimated pharmacokinetic parameters were: Cmax (the observed maximal drug concentration in mg/mL), Tmax (the time for maximal drug concentration in hours), AUC0-8 (the area under the curve was calculated using the trapezoidal rule from zero time to 8 h) and AUC0-∞ (the area under the curve from zero to infinity was calculated from the following equation:
(2) where C8 is the plasma concentration at 8 h.
In addition, the terminal elimination rate constant (k) and the apparent terminal elimination half-life (t1/2) were estimated in h −1 and h, respectively. The terminal elimination rate constant (k) was estimated by log-linear regression analysis on data visually assessed to be a terminal log-linear phase. The apparent terminal elimination half-life (t1/2) was determined as t1/2 = 0.693/k. The relative bioavailability of CLZ from the prepared ODTs was calculated as a ratio between the AUC0-8 of the ODT (F1) to that of Myofen® capsules (250 mg, EVA PHARMA) and expressed on percentage basis as follows:
(3)
Results of in vivo studies were statistically evaluated using unpaired t-test at 5% level of significance.
Statistical analysis
By using Statistical Package for the Social Sciences (SPSS)® software, the one-way ANOVA and the LSD test were used to show the significant or non-significant difference between formulations at p value of 0.05.
Results and discussion
Weight variation
The weight of different ODTs ranged from (145.023 ± 0.002 mg, F4) to (151.221 ± 0.034 mg, F5). All formulations were within the pharmaceutical specification for weight variation (Pharmacopoeia, Citation2002) ().
Table 2. Physical evaluation of the ODT formulations.
Thickness variation
The average thickness of ODTs was found to be in range (3.416 ± 0.057 mm, F3) to (3.942 ± 0.004 mm, F10) ().
Friability
All ODTs did not break or show any capping during the test except F13, F14 and F15. This might be due to the increased ratio of CLZ powder compared with the co-processed excipient in the tablet (2:1). All tablets showed acceptable friability according to the British Pharmacopeia (Pharmacopoeia, Citation2013), except F7, F8 and F12, which had friability >1% ().
Hardness
The hardness of ODT is usually preferable between 2 and 8 kg (Shafei, Citation1998). The hardness of tablets depends on the compression force and the amount and type of binding agent present. The compression force used was the same for all formulations; therefore the change in hardness values of different ODTs observed in could be attributed to the amount and type of binding agents in the co-process excipients (Ochu, Citation2009). The hardness values for all tested tablets were within (2.532 ± 0.213 kg, F15) and (5.724 ± 0.340 kg, F1). So this relatively low hardness provided enough strength and porosity to ensure rapid wetting and disintegration of the tablets. It was clear from the results that the increase of the drug ratio to excipients led to a decrease in the ODT’s hardness. The hardness of ODTs prepared by Pharmaburst®500 (F1 with drug: excipient ratio 0.5:1) was 5.724 ± 0.340 kg, whereas the hardness of F6 (1:1) and F11 (1:0.5) were 4.343 ± 0.149 kg and 3.951 ± 0.229 kg, respectively. This was due to the fact that, the co-excipients had better compression properties compared with CLZ powder. So, increasing the drug to excipients ratio, might have affected the internal structure of the tablet leading to the formation of more fragile tablets.
Drug content
Results in showed that the average drug content of three tablets from each formula ranged from (92.654 ± 3.776%, F9) to (104.54 ± 1.506%, F2) of the label claim. Thus, all formulations complied with the pharmacopeial limits (Pharmacopoeia, Citation2007).
Wetting time
WT is an important parameter for disintegration properties of the tablets (Velmurugan & Vinushitha, Citation2010). Wetting is closely related to the internal structure of tablet and to the hydrophilicity of the excipients (Abdelbary et al., Citation2009). From the results of WT shown in , it was found that all formulations prepared by Pharmaburst®500 (F1, F6 and F11) gave acceptable results (18.83 ± 0.340, 40.48 ± 0.6390, and 45.326 ± 0.938 s), respectively. But in the case of Pearlitol flash® based formulations, only (F2 and F7) gave acceptable results (79.236 ± 0.931 and 171.72 ± 3.124 s), respectively, whereas F12 did not give acceptable results (>180 s). Also in the case of Starlac® based formulations, only F3 gave acceptable result (115.163 ± 1.671 s), but F8 and F13 did not give acceptable results (both >180 s). All Prosolv® odt based formulations did not give acceptable results (>180 s). Finally, all formulations prepared by F-melt® showed acceptable WT results 56.75 ± 1.241, 86.312 ± 0.041, 162.823 ± 0.015 s for F5, F10 and F15, respectively.
By comparing different types of co-processed superdisintegrants-based formulation at the same drug to excipient ratio, it could be shown that Pharmaburst®500 based formulations had the shortest WT. This was attributed to the difference in the type of superdisintegrant included in the different excipients and capacity of these excipients to uptake water (Tayel, Citation2013). Pharmaburst®500 contains crospovidone as superdisintegrant, whereas Pearlitol flash® and Starlac® contain maize starch. Previous researchers showed that crospovidone had higher equivalent force parameter values than that of maize starch (Caramella et al.,Citation1990). Therefore, the rate of water uptake of crospovidone was higher, thus showing faster WT.
Although both Pharmaburst®500, Prosolv® odt and F-melt® consist of crospovidone as superdisintigrant, the results showed they had different WT. In fact, all formulations prepared by Prosolv® odt were not successful and showed prolonged WT. This was attributed to the complex structure of Prosolv® odt as it contained additional microcrystalline cellulose (MCC) and fructose which might lead to increase WT dependent on the applied compression force, so increasing compression force led to increase in WT (Stoltenberg & Breitkreutz, Citation2011). Sunada and Bi (Citation2002) studied the difference between WT of tablets containing MCC and L-HPC. Their results showed that tablets containing more MCC showed lower porosity, as MCC is easily compressible. Moreover, MCC has a smaller particle diameter compared with L-HPC, which is almost completely fibrous, so the number of contact points per unit cross-sectional area of the fracture plane of an MCC tablet is supposed to be larger than that of L-HPC. Additionally, MCC contains more free hydroxyl groups than L-HPC (some of which are substituted by hydroxypropyl groups), and thus the interaction force in a contact point is stronger in case of MCC compared with L-HPC, because the hydrogen bond of hydroxyl groups is considered to be stronger than that of hydroxypropyl groups. In general, the wetting time generally decreases with an increased MCC content, When the MCC content exceeded 90%, however, the wetting time showed a reverse tendency. This suggested that the inner structure of these tablets underwent some change at a high MCC concentration (Sunada & Bi, Citation2002).
In case of F-melt® which also contains crospovidone, previous researchers(Mrhar & Baumgartner, Citation2008), found that the specific surface area of the Pharmaburst®500 is larger than that of F-melt® and it was hypothesized that it could have an impact on either the compression behavior and wetting time or the disintegration time of the tablets obtained.
On the other hand, Pearlitol flash® and Starlac® both possess the same disintegrants (maize starch) but differ in the type of diluent used. Pearlitol flash® has mannitol as diluent, whereas Starlac® has lactose as diluent. Lactose is known to form slowly disintegrating tablets, because it is slow in wetting leading to formation of less porous tablets. Referring to a previous study (Selkirk & Ganderton, Citation1970), where tablets formed of lactose were compared with those formed of sucrose, lactose containing tablets showed lower percentage of pores when measured using mercury porosimetry. So the lactose containing tablets contain less pores and this led to increase in wetting time.
Regarding tablets prepared by Pharmaburst®500, the WT of the prepared tablets decreased by increasing the ratio of Pharmaburst®500. Our results showed that, the WT of formulations F1, F6 and F11 were (18.83 ± 0.340, 40.48 ± 0.6390 and 45.326 ± 0.938 s), respectively. This means that increasing the disintegrant concentration led to a slower WT. So, by increasing the Pharmaburst®500 concentration in the tablet, the effect of crospovidone would be increased and the hindering effect of other components (binding agents, diluents, etc.) on WT might be less.
Results showed that the WT of Pearlitol flash® based formulations decreased by increasing ratio of the excipient in it. The WT of F2 (79.236 ± 0.931 s) was shorter than that of F7 (171.72 ± 3.124 s) and F12 (>180 s). Similarly, Starlac® based formulations showed a decrease in the WT by increasing its concentration. From , we observed that the WT of F3 (115.163 ± 1.671 s) was shorter than that of F8 (>180 s) and F13 (>180 s). Also, WT of ODTs prepared by F-melt® decreased by increasing its concentration from 56.75 ± 1.241 s in case of F5 to 86.312 ± 0.041 and 162.823 ± 0.015 s for F10 and F15, respectively.
In vitro disintegration time
Slow or incomplete disintegration of tablets leads to low bioavailability of the drug from it. Although the compedial standards (Pharmacopeia, Citation2009) state that DT for fast dissolving tablets should be within 3 min, many critics find that a maximum disintegration time of 3 min for any tablet is too long and that the presence of a gritty tablet in the patient’s mouth for 3 min would be unpleasant and uncomfortable (Shoukri et al., Citation2009). According to the literature, the oral disintegration time of mouth-dissolved tablets is 1 min or less, preferably about 30 s or less (Kuno et al., Citation2005). Disintegration depends on the effect of disintegrants and water soluble excipients in the formula (Mizumoto et al., Citation2005).
It was observed that Pharmaburst®500 proved to be the best excipient exhibiting the shortest DT (). This could be explained, as mentioned before, by the higher capacity of crospovidone as superdisintegrant (Caramella et al.,Citation1990), as it had rapid capillary activity and pronounced hydration with little tendency to gel formation, These factors suggest that the DT can be decreased by using wicking type of disintegrants as crospovidone (Gohel et al., Citation2004).
Our results revealed that the shortest DT was observed by F1 (26.434 ± 1.693 s) which contained Pharmaburst®500 in high concentration, and there was a significant difference in DT for Pharmaburst®500 based formulations which contained less amount of the superdisintegrant, as DT for F6 was (28.0 ± 3.464 s) and that of F11 was (43.333 ± 1.527 s).
The most obvious parameter to determine the disintegration performance is the rate of force development (Colombo et al., Citation1984). So, crospovidone showed significantly higher force equivalent values when compared with maize starch; crospovidone was considered to be the best among all disintegrants used in this study (Caramella et al., Citation1990).
Pearlitol flash® based ODTs showed slower DT than those containing Pharmaburst®500 because, in addition to the above reasons, the Pearlitol flash® based formulations showed a non-wetting, hard central core that delayed the DT. Similar results were observed by Jacob et al. (Citation2007), who found that although MCC and mannitol have been extensively used in the formulation of ODTs, the non-wetting property of the hard compact central core might delay the disintegration time.
Similar to the Pharmaburst®500 based formulations, we can arrange the Pearlitol flash® based formulations according to DT in the following ascending order: (F2 < F7 < F12). DT of F2 (35.0 ± 1.0 s) and F7 (67.666 ± 8.082 s) were significantly shorter than F12 (>180 s), which exceeded 3 min. This might be due to the change in the concentration of Pearlitol flash® in different formulations.
Starlac® based formulations showed slower DT than those prepared using Pearlitol flash®, although both Starlac® and Pearlitol flash® contained the same type of superdisintegrant (maize starch). Pearlitol flash® has mannitol as diluent, whereas Starlac® has lactose as diluent. This was the reason for the delay in DT, as observed by Shu et al. (Citation2002). DT of Starlac® based formulations could be arranged in the following ascending order: (F3 < F8 < F13). DT of F3 (50.0 ± 2.530 s) and F8 (84.0 ± 3.605 s) were significantly shorter than that of F13, which had DT that exceeded 3 min.
With respect to all formulations containing Prosolv® odt, we noticed that they had DT more than 3 min. Prosolv® odt consists of crospovidone, MCC, in addition to mannitol which was likely to be the cause of delay of the DT. Moreover, MCC lowered water uptake into the tablet and thus prolonged the DT of the tablet.
Finally, in case of F-melt® which also contains crospovidone, but has specific surface area less than Pharmaburst®500 as mentioned before which was the reason for delay of DT in formulations containing F-melt®. DT of F-melt® based formulations can be arranged in the following ascending order: 64.102 ± 0.986, 105.730 ± 0.061 and 176.436 ± 0.23 s for F5, F10 and F15, respectively.
In vitro dissolution studies
Tablet disintegration is necessary for ODTs, but dissolution is the most important parameter for drug absorption (Zhao & Augsburger, Citation2005).
) represents the in vitro release profile of CLZ from different ODTs formulations. The amount of CLZ dissolved after 15 min (Q15) was used as a parameter to compare different ODT formulations (). The data showed that F1 attained the highest (Q15) value (100.633 ± 0.172%), whereas F9 showed the lowest (Q15) value (20.179 ± 1.474%). The results were in accordance with those of WT and DT. In case of tablets prepared using Pharmaburst®500 (F1, F6 and F11), it was found that formulations containing the highest amount of Pharmaburst®500 (F1) gave higher percent of drug released (100.633 ± 0.172%) compared with F6 (97.408 ± 0.118%) and F11 (79.388 ± 3.393%), which had less amount of Pharmaburst®500 compared with F1 (). It was concluded that the increase in disintegrant concentration resulted in increase in percentage of drug released as mentioned before.
Figure 1. In vitro dissolution profile of CLZ from different ODTs prepared by: (a) Pharmaburst®500; (b) Pearlitol flash®; (c) Starlac®; (d) Prosolv® odt and (e) F-melt®.
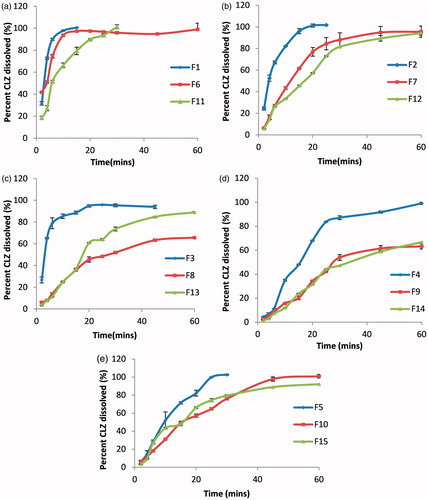
Table 3. Percent of CLZ dissolved after 15 min (Q15) of different ODTs.
In case of formulations prepared using Pearlitol flash® (F2, F7 and F12), it was found that formulations containing the highest amount of Pearlitol flash® (F2) gave higher percent of drug released (96.026 ± 2.363%) than both F7 (61.558 ± 0.357%) and F12 (45.503 ± 0.716%), which had less amount of Pearlitol flash®. ()
In case of formulations prepared by Starlac® (F3, F8 and F13), it was found that formulations containing the highest amount of Pearlitol flash® (F3) gave higher percent of drug released (88.684 ± 1.417%) than F8 (35.941 ± 0.668%) and F13 (37.139 ± 0.394%), which had less amount of Starlac® compared with F3. ()
Prosolv® odt based formulations showed less percent of drug dissolved compared with other formulations. This can be explained by the slow WT and DT as a result of prementioned reasons regarding the composition of Prosolv® odt ().
F-melt® based formulations F5, F10 and F15 showed prolonged dissolution 71.428 ± 1.264%, 47.308 ± 1.772% and 49.961 ± 1.983%, respectively, compared with Pharmaburst®500 because of slow DT as a result of specific surface area as mentioned previously ().
Differential scanning calorimetry
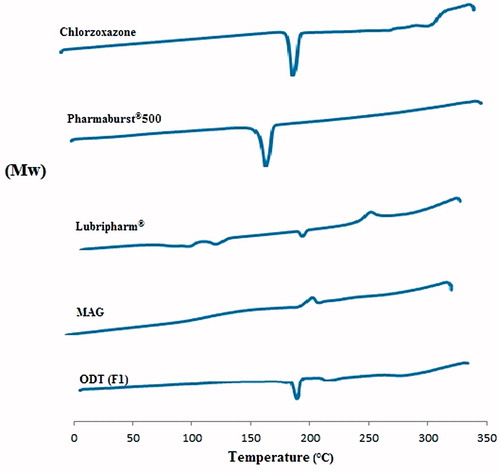
shows the DSC thermogram of pure CLZ, Pharmaburst® 500, MAG, Lubripharm® as well as optimum ODT selected (F1). CLZ showed an endothermic peak at 191.4 °C corresponding to its melting point. The sharp endothermic peak signified that CLZ was in pure crystalline state. The thermogram of Pharmaburst®500 showed an endothermic peak at 166.59 °C due to the mannitol base of the co-processed excipient (the melting point of pure mannitol ranges from 166 to 168 °C). The thermogram of the chosen ODT (F1) did not show the appearance of new peaks, thus indicating no interaction between the drug and the used excipients. However, there was a slight shift in the peak produced by virtue of mixing of components, where the process of mixing was found to reduce the purity of the components (Tayel, Citation2013).
Figure 2. DSC thermograms of CLZ, Pharmaburst® 500, MAG, Lubripharm® as well as selected ODT (F1).
In vivo bioavailability
The HPLC chromatogram of rabbit plasma spiked with CLZ is shown in . The mean plasma concentration–time profiles of CLZ following administration of single dose of ODT (50 mg, F1) to six rabbits and a single oral dose of the reference CLZ capsules (250 mg, Myofen®) to another six rabbits are shown in . The mean pharmacokinetic characteristics are summarized in . After administration of ODT (F1), CLZ was rapidly absorbed and reached a Cmax of 4.169 ± 1.302 μg/mL at a Tmax of 0.33 ± 0.129 h. After the oral administration of Myofen® capsules, CLZ reached a Cmax of 11.043 ± 0.872 μg/mL at a Tmax of 1.083 ± 0.343 h. Regarding the extent of drug absorption as indicated by the AUC0-∞ values, the mean AUC0-∞ was 4.734 ± 0.837 μg.h/mL in ODT (F1) compared with 16.793 ± 3.534 μg.h/mL in Myofen® capsules. The percentage relative bioavailability of ODT (F1) was found 140.95%. The pharmacokinetic parameters AUC0-8, AUC0-∞, Cmax and t1/2 determined for the ODT (F1) were significantly different (p < 0.05) from those of Myofen® capsules. The mean Tmax estimated from the ODT (F1) was significantly shorter (p = 0.0034) compared with that of Myofen® capsules. This faster rate of CLZ absorption obtained from ODT might be attributed to the partial absorption of drug through the membrane of oral cavity and esophagus resulted in the reduction of hepatic first-pass effect of CLZ. Moreover, the rapid absorption of drug from ODT (F1) correlates well with the results of in vitro dissolution, which showed that CLZ was rapidly dissolved from ODT (F1). An earlier bioequivalence study was performed by Zhang & Xu (Citation2003) in human volunteers comparing the formulated CLZ tablets (200 mg/tablet) to orally administered CLZ tablets (200 mg/tablet) on the Chinese market provided by Zhejiang Yatai Pharmaceuticals (batch No.940405). The results of the study proved that the two treatments (reference and test) were bioequivalent and both attained Cmax of 8.41 and 10.3 μg/mL, respectively.
Figure 3. HPLC chromatogram of rabbit plasma spiked with CLZ.
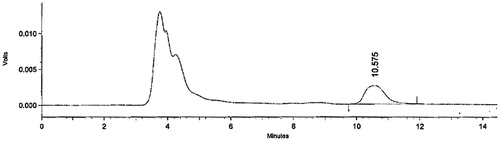
Figure 4. Mean plasma concentration of CLZ following the administration of ODT (F1) (50 mg) and oral administration of Myofen® capsule (250 mg) to 12 rabbits.
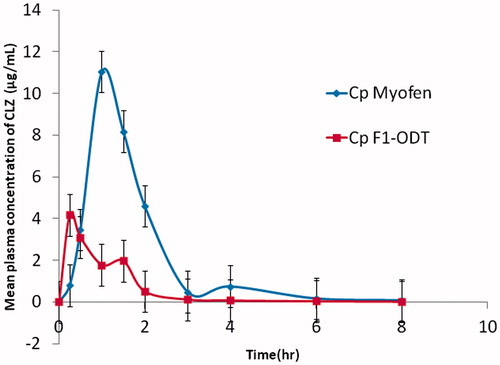
Table 4. Mean pharmacokinetic parameters of CLZ following the administration of ODT (F1) (50 mg) and oral administration of Myofen® capsule (250 mg) to 12 rabbits.
Conclusion
In this study, ODTs of CLZ were successfully prepared by direct compression method using different co-process superdisintegrants. Increasing the ratio of CLZ to co-process superdisintegrants from 0.5:1 to 1:1 and to 2:1, resulted in a significant increase in both the WT and DT. ODT (F1), which contained Pharmaburst® 500 in the highest concentration, showed the shortest DT and WT and possessed the highest Q15min. The in vivo study showed that the Tmax of ODT (F1) was significantly shorter than that of the reference with a relative bioavailability of 140.95%. Based on these findings, it could be concluded that orodispersible tablets are a promising carrier for CLZ designed for management of muscle spasm due to the enhanced dissolution and rapid absorption of the drug through the oral mucosa. Orodispersible tablets of CLZ could be considered a pharmaceutical alternative for conventional oral capsules helping in fast alleviation of muscle spasm and decreasing the dose of the drug with subsequent advantages of lessening the cost and side effects. Further clinical studies on the developed ODTs are recommended to be performed to assess the bioavailability of CLZ ODTs in humans.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Abdelbary A, Elshafeey AH, Zidan G. (2009). Comparative effects of different cellulosic-based directly compressed orodispersable tablets on oral bioavailability of famotidine. Carbohyd Polym 77:799–806
- Al-Khattawi A, Mohammed AR. (2013). Compressed orally disintegrating tablets: excipients evolution and formulation strategies. Expert Opin Drug Deliv 10:651–63
- Alli SMA. (2011). Orally disintegrating drug delivery systems: a technical note on technologies and issues. Int J Pharm Front Res 1:38–55
- Bolhuis GK, Zuurman K, Wierik Te GHP. (1997). Improvement of dissolution of poorly soluble drugs by solid deposition on a super disintegrant. II. The choice of super disintegrants and effect of granulation. Eur J Pharm Sci 5:63–9
- British Pharmacopoeia (2007). The stationary Office under license from the controller of Her Majestys Stationary office for the department of health: A11, A112, A153
- British Pharmacopoeia. (2013) . Published by The Stationery Office on behalf of the Medicines and Healthcare products Regulatory Agency (MHRA), London
- Caramella C, Ferrari F, Bonferoni MC, Ronchi M. (1990). Disintegrants in solid dosage forms. Drug Dev Ind Pharm 16:2561–77
- Chang RK, Xiaodi G, Burnside BA, Couch RA. (2000). Fast-dissolving tablets. Pharm Tech 24:52–8
- Chou R, Peterson K.Helfand M. (2004). Comparative efficacy and safety of skeletal muscle relaxants for spasticity and musculoskeletal conditions: a systematic review. J Pain Symptom Manage 28:140–75
- Colombo P, Ubaldo C, Caramella C, et al. (1984). Disintegrating force as a new formulation parameter. J Pharm Sci 73:701–5
- Dixit RP, Puthli SP. (2009). Oral strip technology: overview and future potential. J Control Release 139:94–107
- European Pharmacopeia. (2009). Sixth edition, published by Council of Europe, Stasbourge, France: 1169–70
- European Pharmacopoeia (2002). Published by the directorate for the quality of medicines of the council of Europe 9EDQM, Strasbourg, France
- Florey K, Al-Badr AA, Brenner Gerald S. (1987). Analytical profiles of drug substances. London: Academic Press Inc., 137
- Gohel M, Patel M, Amin A, et al (2004). Formulation design and optimization of mouth dissolve tablets of nimesulide using vacuum drying technique. AAPS Pharm Sci Tech 5:10–15
- Heer DA, Geeta A, Kumar SH. (2013). Recent trends of fast dissolving drug delivery system–an overview of formulation technology. Pharmacophore 4:1–9
- Honey G, Parshuram R, Rana V, Tiwary AK. (2008). Orally disintegrating systems: innovations in formulation and technology. Recent Pat Drug Deliv Formul 2:258–74
- Jacob S, Shirwaikar AA, Joseph A, Srinivasan KK. (2007). Novel co-processed excipients of mannitol and microcrystalline cellulose for preparing fast dissolving tablets of glipizide. Ind J Pharm Sci 69:633
- Kuno Y, Kojima M, Ando S, Hiroaki N. (2005). Evaluation of rapidly disintegrating tablets manufactured by phase transition of sugar alcohols. J Control Release 105:16–22
- Mizumoto T, Masuda Y, Yamamoto T, et al (2005). Formulation design of a novel fast-disintegrating tablet. Int J Pharm 306:83–90
- Mrhar A, Baumgartner S. (2008). 7th Central European Symposium on Pharmaceutical Technology and Biodelivery Systems, September 18-20, 2008, Hotel Mons, Ljubljana, Slovenia: Proceedings from the Symposium, Slovenian Pharmaceutical Society
- Nagaraju T, Gowthami R, Rajashekar M, Sandeep S, Mallesham M, Sathish D, Shravan Kumar Y. (2013). Comprehensive review on oral disintegrating films. Curr Drug Deliv 10:96108
- Ochu SN. (2009). Evaluation of barley (Hordeum vulgare) starch as a tablet excipient. MSc Thesis submitted to Ahmadu Bello, University of Zaria, Nigeria
- Patel M, Patel N, Patel M. (2007). Fast-dissolving rofecoxib tablets: formulation development & optimization using factorial design. Drug Del Technol 7:33–8
- Sammour OA, Hammad Mohammed A, Megrab Nagia A, Zidan Ahmed S. (2006). Formulation and optimization of mouth dissolve tablets containing rofecoxib solid dispersion. AAPS PharmSciTech 7:E167–75
- Seager H. (1998). Drug-delivery products and the Zydis fast-dissolving dosage form. J Pharm Pharmacol 50:375–82
- Selkirk AB, Ganderton D. (1970). An investigation of the pore structure of tablets of sucrose and lactose by mercury porosimetry. J Pharm Pharmacol 22:79S–85S
- Shafei AH. (1998). Master Thesis, Cairo University, Cairo, Egypt, 51
- Shoukri RA, Iman SA, Shamma RN. (2009). In vitro and in vivo evaluation of nimesulide lyophilized orally disintegrating tablets. Eur J Pharm Biopharm 73:162–71
- Shu T, Hideshi S, Kenji H, Ito K. (2002). Studies of rapidly disintegrating tablets in the oral cavity using co-ground mixtures of mannitol with crospovidone. Chem Pharm Bull 50:193–8
- Stoltenberg I, Breitkreutz J. (2011). Orally disintegrating mini-tablets (ODMTs)-a novel solid oral dosage form for paediatric use. Eur J Pharm Biopharm 78:462–9
- Sunada H, Bi Y. (2002). Preparation, evaluation and optimization of rapidly disintegrating tablets. Powder Technol 122:188–98
- Suresh S, Pandit P, Joshi HP. (2007). Preparation and evaluation of mouth dissolving tablets of salbutamol sulphate. Ind J Pharm Sci 69:467
- Sweetman SC, Pharm B, PharmS FR. (2009). Martindale: the complete drug reference. 36th ed. London, UK: The Pharmaceutical Press, an imprint of RPS Publishing
- Tayel SA. (2013). Pharmaceutical study on an antimigraine drug. Ph.D thesis submitted from Mohammed Hassan, Cairo University, Cairo, Egypt, 107–20
- Velmurugan S, Vinushitha S. (2010). Oral disintegrating tablets: an overview. Int J Chem Pharm Sci 1:1–12
- Wilson CG, Washington N, Peach J, et al (1987). The behaviour of a fast-dissolving dosage form (Expidet) followed by γ-scintigraphy. Int J Pharm 40:119–23
- Xu WJ, Xie H-J, Cao QR, et al (2014). Enhanced dissolution and oral bioavailability of valsartan solid dispersions prepared by a freeze-drying technique using hydrophilic polymers. Drug Deliv 23:41–8
- Zhang S-J, Xu Wei R. (2003). Study on bioequivalence of chlorzoxazone tablets in Chinese volunteers. Asi J Drug Metab Ph 4:53–6
- Zhao N, Augsburger LL. (2005). Functionality comparison of 3 classes of superdisintegrants in promoting aspirin tablet disintegration and dissolution. AAPS PharmSciTech 6:E634–40