
Abstract
Context: Self-emulsifying drug delivery systems (SEDDS) are among most promising tools for improving oral peptide bioavailability.
Objective: In this study, in vitro protective effect of SEDDS containing desmopressin against presystemic inactivation by glutathione and α-chymotrypsin was evaluated.
Materials and methods: The partitioning coefficient (log P) of desmopressin was increased via hydrophobic ion pairing using anionic surfactants. Solubility studies were performed to select the appropriate solvents for SEDDS preparation. Subsequently, droplet size and emulsification properties of 22 SEDDS formulations were evaluated. Moreover, the peptide-surfactant complex was dissolved in two chosen SEDDS formulations. Finally, SEDDS containing desmopressin were characterized regarding lipase stability, toxicity, and in vitro protective effect toward glutathione and α-chymotrypsin.
Results: Desmopressin log P was increased from initial −6.13 to 0.33 using sodium docusate. The resulting desmopressin docusate complex (DES/AOT) was incorporated in two different SEDDS formulations, containing Capmul 907 P as main solvent. DES/AOT-SEDDS-F4 (containing 0.07% w/w DES/AOT) was composed of 50% Capmul 907P, 40% Cremophor RH40, and 10% Transcutol. The comparatively more hydrophilic formulation DES/AOT-SEDDS-F15 (containing 0.25% w/w DES/AOT) consisted of 20% Capmul 907P, 40% Acconon MC8-2, and 40% Tween 20. Both formulations were stable toward digestion by lipase and protected desmopressin toward α-chymotrypsin degradation. Moreover, DES/AOT-SEDDS-F4 also protected the peptide from thiol/disulfide exchange reactions with glutathione and was not cytotoxic at a concentration of 0.375% (w/w).
Conclusion: DES/AOT-SEDDS-F4 protected desmopressin from in vitro glutathione and α-chymotrypsin degradation. DES/AOT-SEDDS-F4 was metabolically stable and nontoxic. Therefore, it could be considered as a potential delivery system for oral desmopressin administration.
Introduction
Oral peptide delivery still remains one of the ultimate challenges in the pharmaceutical–technological field (Fosgerau & Hoffmann, Citation2015; Wang et al., Citation2015). Orally administered peptides are facing various obstacles concerning the ADME process. For instance, peptides must avoid acidic or enzymatic degradation, pass mucus barriers and, in case of drugs containing one or more disulfide bonds, also the so-called sulfhydryl barrier (Leonaviciute & Bernkop-Schnürch, Citation2015). Human physiology with highly acidic conditions in the stomach, secreted enzymes like trypsin, α-chymotrypsin, and elastase along with poor intestinal membrane permeability contribute to an overall low oral peptide bioavailability (Wang et al., Citation2015). However, some oral peptide formulations are yet available on the market (Shen, Citation2003; Fosgerau & Hoffmann, Citation2015; Wang et al., Citation2015), whereby peptide drugs like vancomycin (Vancocin®) and linaclotide (Linzess®) are targeted only for local intestinal delivery. In contrast, the lipophilic peptide cyclosporine (Neoral®) and the highly potent vasopressin structural analog desmopressin (DDAVP®, DesmoMelt®, Minirin®, Octim®, Stimate®) exhibit sufficient systemic effects after oral administration (Wang et al., Citation2015).
Desmopressin is used in the treatment of diabetes insipidus, primary nocturnal enuresis, hemophilia A, Type I von Willebrand’s disease and also to improve human memory function (Wang et al., Citation2002). It is a hydrophilic molecule with a relative high molecular weight of 1069 Da rendering it poorly absorbed from intestinal mucosa. Additionally, it contains a disulfide bond and a cleavage site for α-chymotrypsin. This results in an extensive presystemic degradation and peptide inactivation via α-chymotrypsin and sulfhydryl compounds after oral administration (Kahns et al., Citation1993; Fredholt et al., Citation1999; Wang et al., Citation1999,Citation2015). Consequently, the oral bioavailability of the above mentioned formulations is less than 1% (Kahns et al., Citation1993; Ilan et al., Citation1996; Fredholt et al., Citation1999; Wang et al., Citation2015). Thus, several attempts to develop alternative oral delivery systems such as mucoadhesive submicron emulsions (Ilan et al., Citation1996), solid lipid particles (Christophersen et al., Citation2014) as well as prodrugs (Kahns et al., Citation1993; Wang et al., Citation1999,Citation2002) have been undertaken to increase its bioavailability.
Self-emulsifying drug delivery systems (SEDDS) are undoubtedly among the most promising tools for innovative oral peptide delivery. SEDDS are anhydrous mixtures of oils, surfactants, co-surfactants, and co-solvents, whereby the preconcentrate usually includes the dissolved drug. Upon contact with gastrointestinal fluids, SEDDS spontaneously form stable emulsions containing the drug within oily droplets. Accordingly, the drug is protected against the harsh gastrointestinal environment in the human body (Christiansen et al., Citation2014). In previous studies, it was shown that the model peptide leuprolide is protected against degradation by proteases after incorporation into SEDDS (Hintzen et al., Citation2014). In addition, certain SEDDS can also be attributed with mucus-permeating properties (Friedl et al., Citation2013) and stability toward pancreatic lipase (Mahjub et al., Citation2015). Taking this into consideration, SEDDS may also provide protection against presystemic inactivation of desmopressin. However, to the best of our knowledge, no SEDDS protecting desmopressin against α-chymotrypsin and unintended thiol disulfide interactions have been designed yet.
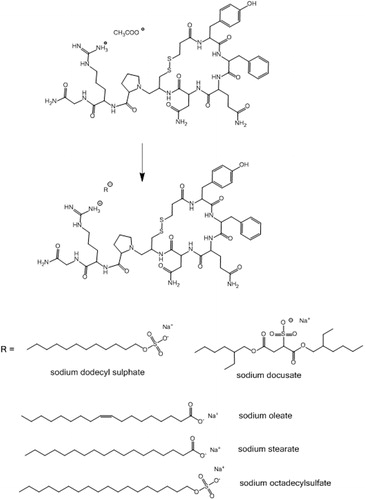
It was therefore the aim of this study to design SEDDS for oral desmopressin delivery protecting the peptide toward aforementioned potential presystemic inactivation via α-chymotrypsin and glutathione. Desmopressin’s lipophilicity was altered by a straightforward hydrophobic ion pairing method (Meyer & Manning, Citation1998) as illustrated in . The desmopressin-surfactant complex was incorporated in SEDDS, which were characterized regarding droplet size, cytotoxicity and stability against pancreatic lipase, reduced glutathione, and α-chymotrypsin degradation.
Figure 1. Anionic surfactants capable of precipitating desmopressin from aqueous solution. The surfactants likely formed an ion pair with the positively charged guanidine group.
Materials and methods
Materials
Desmopressin acetate was purchased from Jinlan Pharm-Drugs Technology Co., Limited (China), Dermofeel BGC, Dermofeel TC-7, Dermofeel MCT were a gift from Dr. Streatmans (Germany), Captex 300, Capmul MCM EP, Capmul 907P, Capmul PG-8, Capmul PG-12, Acconon MC8-2 EP/NF and Caprol 3GO were a gift from Abitec (USA), Labrafil M 1944 CS, Peceol and Transcutol HP were a gift from Gattefossé (France), Mygliol 840 was purchased from Sasol (Germany), sodium stearate and sodium docusate were purchased from Alfa Aesar (Germany), Cremophor RH40, Tween 20, Tween 80, sodium oleate, sodium dodecyl sulfate, sodium octadecylsulphate, reduced glutathione, α-chymotrypsin (from bovine pancreas, Type II, 96 units/mg protein), lipase from porcine pancreas (Type II, 54 units/mg using triacetin), bile salts (1:1 mixture of sodium cholate and sodium deoxycholate) were purchased from Sigma Aldrich (Germany). All other chemicals, reagents and solvents were of analytical grade and received from commercial sources.
HPLC analysis
Desmopressin was quantified on a Hitachi EliteLaChrom HPLC-System equipped with L-2130 pump, L-2200 autosampler and L-2450 photodiode array UV detector. A Nucleosil 100-5 C18 column (250 × 4 mm, 5 μm) was used as stationary phase. A binary solvent system was used at room temperature and flow rate of 1 mL/min. Solvent A was acetonitrile and solvent B was 0.1% (v/v) trifluoroacetic acid (TFA) in water. An isocratic method (25% solvent A) or a gradient method, where 0–6 min: 25% A, 6–8 min: 25–90% A, 8–15 min: 90% A, 15–20 min 90–25% A and 20–26 min: 25% A were used as mobile phase. The general method for washing the column was as follows: 0–30 min: 25–90% A, 30–50 min: 90% A, 50–100 min: 90–60% A, and 100–120 min: 60% A.
Preparation of desmopressin-surfactant complex
Surfactant solutions (50 μL) in molar ratios 0.5–3.0, namely sodium dodecyl sulfate (0.27–1.62 mg/mL), sodium oleate (0.28–1.68 mg/mL), sodium stearate (0.29–1.74 mg/mL), sodium docusate (0.42–2.52 mg/mL), and sodium octadecyl sulfate (0.35–2.10 mg/mL) with respect to desmopressin were slowly added in a dropwise manner to desmopressin solution (50 μL, 2 mg/mL) at room temperature. The precipitated desmopressin-surfactant complex formed was isolated by centrifugation (5 min, 10 000 rpm). The precipitate was washed with purified water, lyophilized (Christ Gamma 1-16 LSC Freeze dryer) and stored at −20 °C for further use. The amount of precipitated desmopressin-surfactant complex was calculated by measuring the remaining amount of peptide in the supernatant by HPLC as described above using EquationEquation (1)(1) :
(1)
where Cs is desmopressin concentration in supernatant and Ct is the total desmopressin concentration.
Partitioning coefficient log P (Capmul 907P/water)
Surfactant solutions (50 μL) in molar ratios 1.0–2.0, namely sodium dodecyl sulfate (0.54–1.08 mg/mL), sodium oleate (0.56–1.12 mg/mL) and sodium docusate (0.84–1.68 mg/mL) with respect to desmopressin were slowly added in a dropwise manner to desmopressin solution (50 μL, 2 mg/mL) to precipitate the peptide-surfactant complex. After 5 min 100 μL of Capmul 907 P was added and stirred for 30 min (900 rpm, 25 °C). The separation of aqueous and Capmul 907 P phase was achieved by centrifugation (5 min, 10 000 rpm). The amount of desmopressin in both phases was determined by HPLC as described above. 10 μL of Capmul 907 P samples were diluted prior to injection in HPLC with 90 μL of isopropanol and 50 μL of deionized water to yield a final volume of 150 μL.
Solubility studies
Desmopressin acetate (0.25 mg) and DES/AOT (0.25 mg) were dispersed in 50 mg of water insoluble oils and water insoluble surfactants (HLB < 12) as listed in . The dispersion was homogenized by sonication for 2 min and placed on a thermomixer (25 °C, 1000 rpm) for 24 h. Separation of undissolved peptide and oil was achieved by centrifugation (5 min, 18 000 rpm). 10 mg of oil was mixed with 60 μL of isopropanol and homogenized. Next, 140 μL of deionized water was added to precipitate the oil. Complete phase separation was induced by centrifugation (10 min, 18 000 rpm). The amount of desmopressin in the separated aqueous phase was determined by HPLC as described above.
Table 1. Classification of lipid-based excipients used in the study.
SEDDS development, characterization, and DES/AOT incorporation
Mixtures of oils, surfactants, and co-solvents (1 g) were homogenized by mixing to form a single phase. Cremophor RH40 was melted at 40 °C before use. SEDDS preconcentrates (100 mg) were emulsified in 5 mL of 50 mM phosphate buffer pH 6.8. Mean droplet size and polydispersity index of emulsions were determined by dynamic light scattering using a PSS Nicomp 380 DLS (Santa Barbara, CA). Measurements were performed at 25 °C. DES/AOT was incorporated in SEDDS formulation F4 as follows (DES/AOT-SEDDS-F4): 0.66 mg of DES/AOT was immediately dissolved in 100 mg of Transcutol. Then 500 mg of Capmul 907 P was added and mixed. Finally, 400 mg of Cremophor RH40 was added to the mixture dropwise under stirring (800 rpm). DES/AOT-SEDDS-F4 was homogenized by stirring for additional 20 min at room temperature. DES/AOT was incorporated in SEDDS formulation F15 as follows (DES/AOT-SEDDS-F15): Formulation F15 was prepared by mixing 200 mg of Capmul 907 P, 400 mg of Tween 20 and 400 mg of Acconon MC8-2 at room temperature. Then 2.5 mg of DES/AOT was added to 1000 mg of formulation F15 and left to stir (800 rpm) for 24 h at room temperature. DES/AOT-SEDDS-F4 and DES/AOT-SEDDS-F15 were centrifuged (5 min, 13 400 rpm). No sediment indicated the complete DES/AOT dissolution.
Stability studies
Stability of dissolved DES/AOT in SEDDS was determined as follows: DES/AOT-SEDDS-F4 and DES/AOT-SEDDS-F15 were stored in a sealed container at 25 °C and 40% RH, protected from light. SEDDS were centrifuged (5 min, 13 400 rpm) after 1, 3, 7, 14, and 21 d to visually evaluate if DES/AOT precipitated. Additionally, 10 mg of DES/AOT-SEDDS-F4 and DES/AOT-SEDDS-F15 were emulsified in 1 mL of deionized water. The emulsion was immediately destroyed by addition of 150 mg of NaCl and centrifugation (5 min, 13 400 rpm). The amount of desmopressin in the aqueous phase was determined by HPLC as described above.
SEDDS in vitro digestion study
For SEDDS in vitro digestion studies, a slightly modified method was used as described previously by Fatouros et al. (Citation2007). The composition of digestion medium pH 6.5 was 2 mM Trizma maleate, 150 mM NaCl, 5 mM CaCl2, and 5 mM bile salts. DES/AOT-SEDDS-F4 and DES/AOT-SEDDS-F15 (1% w/v) were emulsified in the digestion medium in a thermostatically controlled vessel (37 °C) and pH was adjusted to 6.5 with 0.1 M HCl or 0.1 M NaOH. Freshly suspended pancreatic lipase in digestion medium (81 U/mL) was immediately added to the medium containing SEDDS to initiate lipolysis. Throughout the study pH was kept constant at 6.5 by adding 0.1 M NaOH. The rate of SEDDS digestion was calculated from NaOH consumption.
Glutathione stability studies
For glutathione stability studies, a slightly modified method was used as described previously by Schmitz et al. (Citation2006). In brief, desmopressin acetate (0.10 mg/mL) and reduced glutathione (5 mg/mL) were dissolved in 50 mM acetate buffer pH 5.5. Separately, 100 mg of DES/AOT-SEDDS-F4 and DES/AOT-SEDDS-F15 were emulsified in 2500 μL of acetate buffer. After 15 min of incubation at 37 °C 700 μL of glutathione (5 mg/mL) was added to 700 μL of samples yielding the final SEDDS concentration of 2% (w/v). At predetermined time points aliquots of 100 μL were withdrawn. The emulsion was destroyed by adding 30 mg of sodium chloride. The complete separation of aqueous and oily phase was achieved by centrifugation (5 min, 14 000 rpm). The amount of remaining intact desmopressin in the aqueous phase was determined by HPLC as described above.
Enzymatic stability studies
Desmopressin acetate (0.12 mg/mL) was dissolved in 50 mM HEPES buffer pH 7.4 containing 2 mM CaCl2. Separately, 10 mg of DES/AOT-SEDDS-F4 and DES/AOT-SEDDS-F15 were emulsified in 500 μL of HEPES buffer. After 15 min of incubation at 37 °C 500 μL of α-chymotrypsin (3.6 mg/mL) was added to the 500 μL of samples yielding the final SEDDS concentration of 1% (w/v). At predetermined time points aliquots of 70 μL were withdrawn and added to 70 μL of 2% TFA in order to quench the enzymatic reaction. The emulsion was destroyed by adding 30 mg of sodium chloride. The complete separation of aqueous and oily phase was achieved by centrifugation (5 min, 14 000 rpm). The amount of remaining intact desmopressin in the aqueous phase was determined by HPLC as described above.
Resazurin assay
The potential cytotoxic effect of 0.25% and 0.375% (w/v) SEDDS formulations was determined by a slightly modified resazurin assay as described previously (Partenhauser et al., Citation2015). Approximately 2.5 × 104 Caco-2 cells per well were seeded to a 24-well plate. The cells were incubated for 14 d in MEM supplemented with 10% (v/v) heat inactivated fetal calf serum (FCS) and penicillin/streptomycin solution (100 units/0.1 mg/L) at 95% humidity and 37 °C in an atmosphere of 5% CO2. Pure MEM and Triton X 100 served as negative and positive controls, respectively. After 1 h and 4 h of incubation, samples (500 μL) were removed from the cells and washed with 250 μL of isotonic phosphate buffered saline. Subsequently, 500 μL of resazurin solution (44 μM) in FCS- and penicillin/streptomycin-free MEM was added to each well and incubated for 3 h. The fluorescence of the supernatant was measured after background subtraction using an excitation wavelength of 540 nm and an emission wave length of 590 nm. Cell viability rates were calculated according to EquationEquation (2)(2) :
(2)
where As is the fluorescence of samples and Ac is the fluorescence measured after treatment of cells with MEM.
Statistical data analysis
Statistical data analysis was performed using the Graph Pad Prism 5 software. ANOVA tests with p < 0.05 as the minimal level of significance followed by Turkey’s multiple comparison test with p < 0.05 as minimal level of significance were performed. All values are expressed as the means ± SD.
Results and discussion
Preparation of desmopressin-surfactant complex
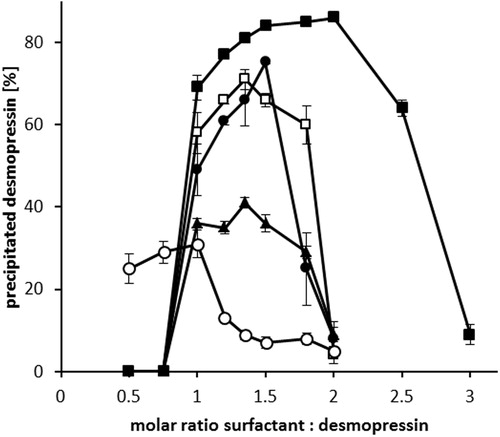
Desmopressin is a cationic peptide with one ionizable guanidine group (pKa ∼12) and an estimated isoelectric point of 10.22 (Wang et al., Citation2015). Aqueous desmopressin acetate solution (2 mg/mL) has a pH value of 4–5, yielding desmopressin a net positive charge. When anionic surfactants such as sodium octadecyl sulfate, sodium dodecyl sulfate, sodium oleate, sodium stearate, and sodium docusate were used as counter ions, desmopressin precipitated from the solution in the form of peptide-surfactant complex (). The formation of ion pair complex is conventionally determined by measuring the UV-Vis absorbance (Choi & Park, Citation2000). However, in this study HPLC determination was the preferred method due to its low limit of quantification and the possibility to directly measure the amount of precipitated peptide. Sodium dodecyl sulfate, sodium oleate, and sodium docusate were more efficient in precipitating desmopressin by more than 70% in comparison to sodium stearate and sodium octadecylsulphate, where the precipitation yield did not exceed 40%. The latter surfactants have a limited aqueous solubility and had to be heated to 60 °C prior to addition to desmopressin solution. The addition of heated surfactant solutions to desmopressin solution (25 °C) might have precipitated the surfactants and lowered their overall amount available for ion pairing. In all cases, at a certain ratio a maximum precipitation was observed demonstrating that both too low and too high amount of added surfactants resulted in lowered yields. This indicates the importance of molar ratio of surfactant (S/D) to desmopressin on peptide precipitation. At low S/D ratios, the precipitation is negligible as the amount of surfactant is likely too low to rise the lipophilic character of the peptide sufficiently. With increasing S/D ratios the excessive surfactant might have solubilized the precipitated peptide complex via micellization (Choi & Park, Citation2000). In most cases, the regained peptide aqueous solubility occurred when S/D ratio exceeded 2:1 except when sodium docusate was used, where the micellization S/D ratio required was 2.5:1.
Figure 2. Hydrophobic ion pairing of desmopressin acetate with sodium octadecyl sulfate (○), sodium stearate (▴), sodium dodecyl sulfate (□), sodium oleate (•), and sodium docusate (▪). After the addition of surfactants, the precipitated desmopressin complex was centrifuged and the amount of remaining peptide in supernatant determined by HPLC. Data are shown as mean ± SD (n = 3).
Partitioning coefficient log P (Capmul 907 P/water)
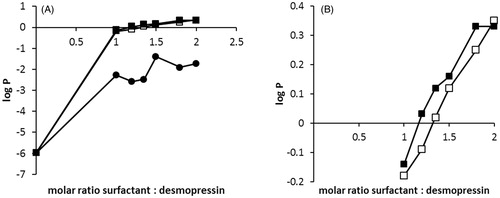
The partitioning coefficient log P (octanol/water) is an indicator of the drugs lipophilicity. Sufficient lipophilicity of the peptide-surfactant complex is crucial for its incorporation in SEDDS and increased cell membrane permeability (Kahns et al., Citation1993). In previous studies, desmopressin acetate and its ester prodrug derivatives were partitioned between 50 mM phosphate buffer pH 7.4 and octanol phase. Neither desmopressin acetate nor desmopressin propionate ester diffused in octanol phase even when octanol: phosphate buffer ratio of 1000:1 was used (Kahns et al., Citation1993). These findings indicated that octanol was a too lipophilic solvent for desmopressin-partitioning studies. In this study, partitioning of the peptide-surfactant complex between aqueous and comparatively more hydrophilic Capmul 907 P phase using three most promising surfactants, namely sodium dodecyl sulfate (SDS), sodium oleate (SOL), and sodium docusate (AOT) was evaluated. Results presented in indicate that desmopressin-sodium oleate (DES/SOL) complex failed to migrate to the Capmul 907 P phase in the tested molar range S/D of 1:1 to 2:1. However, the log P (DES/SOL) was increased to −1.40 compared to the initial calculated desmopressin acetate log P of −6.13 (Wang et al., Citation2015). In contrast, a significant increase (p < 0.05) of log P (DES/SDS) and log P (DES/AOT) to a positive value of 0.33 was observed (). Although a positive log P value was reached, DES/AOT and DES/SDS are still considered hydrophilic in comparison with low lipophilic drugs like nifedipine (log P = 2.20) (Jannin et al., Citation2015) and highly lipophilic drugs like atorvastatin (log P = 6.36) (Belhadj et al., Citation2013), which were already successfully incorporated into SEDDS (Belhadj et al., Citation2013; Jannin et al., Citation2015).
Figure 3. (A) Partitioning coefficient log P (Capmul 907 P/water) of DES/SOL (•), DES/AOT (▪) and DES/SDS (□). Surfactant solutions were added in a dropwise manner to desmopressin solution to precipitate the peptide-surfactant complex. After 5 min 100 μL of Capmul 907 P was added and the mixture stirred for 30 min. The amount of desmopressin in both phases was determined by HPLC. (B) Zoomed log P (Capmul 907 P/water) for DES/AOT (▪) and DES/SDS (□).
Solubility studies
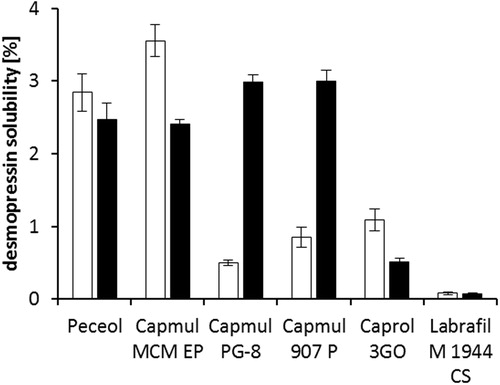
The most efficient desmopressin log P (Capmul 907 P/water) increase was achieved by using sodium docusate in S/D ratio of 1.8:1. Therefore, DES/AOT was chosen for further studies. A comparative solubility study between desmopressin acetate and desmopressin docusate was performed to identify the most appropriate water insoluble oils for SEDDS development. The chosen water insoluble oils and surfactants are listed in . Both desmopressin acetate and DES/AOT were practically insoluble in strong lipophilic solvents like Dermofeel BGC, Dermofeel MCT, Dermofeel TC-7, Captex 300, Mygliol 840, ethyl oleate, and also Capmul PG-12. In contrast, comparatively more hydrophilic solvents were able to solubilize desmopressin (). Peceol and Capmul MCM solubilized similar amounts of both desmopressin salts. Moreover, the solubility of DES/AOT was significantly increased (p < 0.05) compared to desmopressin acetate in Capmul 907 P and Capmul PG-8. As peptide-surfactant complexes may also contain bare peptide (Hintzen et al., Citation2014) a solvent with significant difference in solubility between desmopressin acetate and DES/AOT is desired to guarantee predominantly the dissolution of the more lipophilic form. Furthermore, hydrophilic proteins and peptides have a tendency to migrate into the aqueous phase after emulsification with gastrointestinal fluid (Rao & Shao, Citation2008). Therefore, Capmul 907 P was chosen as oily phase for SEDDS development due to a slightly greater difference in solubility of both desmopressin salts in comparison with Capmul PG-8.
Figure 4. Solubility of desmopressin acetate (white bars) and DES/AOT (black bars) in chosen water insoluble lipids and surfactants after 48 h of incubation at 25 °C. Data are shown as mean ± SD (n = 3).
SEDDS development, characterization, and DES/AOT incorporation
According to abovementioned results Capmul 907 P was chosen as oil for DES/AOT incorporation. Due to relative high DES/AOT hydrophilicity hydrophilic surfactants with high HLB values, namely Tween 20, Cremophor RH40, Tween 80, and Acconon MC8-2 were chosen for SEDDS development (). The chosen surfactants are generally considered as safe, biocompatible, and efficient (Shen & Zhong, Citation2006) – especially Cremophor RH40 (Thomas et al., Citation2012). Additionally, Tween 20 was shown to be a permeation enhancer (Bandivadekar et al., Citation2013). SEDDS containing Capmul 907 P as main solvent are listed in . Formulations with Cremophor RH40 exhibited smaller droplet sizes than those with Tween 80. Moreover, different surfactant ratios of Tween 20 and Acconon MC8-2 were investigated regarding their emulsifying properties Formulations F18–F22 containing 30% Capmul 907 P failed to form emulsions with droplet sizes below 1000 nm, whereas formulations F14–F16 containing 20% Capmul 907 P formed emulsions with droplet sizes in the range of 10–30 nm. The ratio of Tween 20 to Acconon MC8-2 in the surfactant mixture had an impact on droplet size. Formulation F13 containing 20% Tween 20, 60% Acconon MC8-2 and 20% Capmul 907 P formed emulsions with a mean droplet size of 748 nm, whereas at 60% Tween 20, 20% Acconon MC8-2 and 20% Capmul 907 P no droplets were formed. The increasing amounts of Tween 20 resulted in overall decreased droplet size. The higher hydrophilicity of Tween 20 compared to Acconon MC8-2 might have been responsible for effective solubilization of the oily components. Among formulations F1–F12 composed of 40–50% hydrophilic surfactants formulation F4 formed emulsions with the smallest droplet size and low polydispersity index. Furthermore, among the comparatively more hydrophilic formulations F13–F22 composed of 70–-80% hydrophilic surfactants formulation F15 was identified with the narrowest droplet size distribution. Therefore, F4 and F15 were chosen for further studies.
Table 2. SEDDS formulation development with Capmul 907P as main solvent.
Stability studies
A 21-d stability study of DES/AOT-SEDDS-F4 and DES/AOT-SEDDS-F15 demonstrated that DES/AOT remained stable in both formulations under specified conditions. In all cases, no sediment was observed after centrifugation indicating that desmopressin did not precipitate from SEDDS and thus remained in the complex with sodium docusate. Additionally, HPLC showed that after 21 d 98.4% of desmopressin remained intact in DES/AOT-SEDDS-F4 and 97.5% in DES/AOT-SEDDS-F15. Subsequently, DES/AOT-SEDDS-F4 and DES/AOT-SEDDS-F15 formulations could be considered as stable.
In vitro SEDDS digestion study
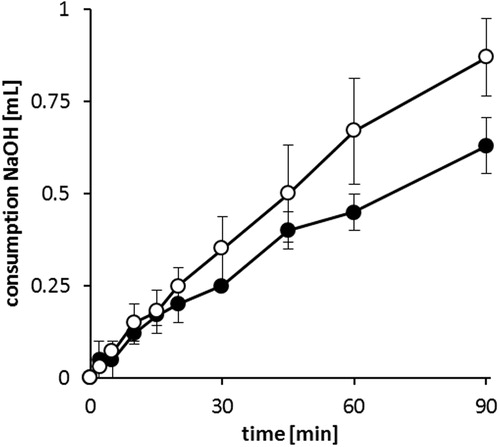
Upon oral administration, SEDDS are prone to digestion in duodenum by pancreatic lipase. It hydrolyzes triglycerides in positions 1 and 3, resulting in two free fatty acids and one 2-monoacylglyceride (Müllertz et al., Citation2010). The consumption of 0.1 M NaOH during lipolysis of formulations F4 and F15 is illustrated in . In 90 min less than 1 mL of 0.1 M NaOH was consumed indicating the relative stability of DES/AOT-SEDDS-F4 and DES/AOT-SEDDS-F15 toward lipase. DES/AOT-SEDDS-F4 contains Cremophor RH40 as triglyceride, which was shown not only to be a weak lipase substrate but also to inhibit the lipolysis process (Fatouros et al., Citation2007). Other SEDDS components containing ester bonds include monoesters Capmul 907 P, Tween 20 and Acconon MC8-2, which is a mixture of monoesters, diesters, and triesters of glycerol, and monoesters and diesters of polyethylene glycols. Low NaOH consumption may be due to lack of ester bonds or simply the fact that the aforementioned components are weak lipase substrates. For comparison, under the same experimental conditions NaOH consumption by SEDDS containing high amount (65% w/w) of medium chain triglycerides (tricaprylin and Capmul MCM) was around 5 mL (Zupančič et al., Citation2016).
Figure 5. Consumption of 0.1 M NaOH during in vitro digestion of SEDDS formulations F4 (○) and F15 (•) by pancreatic lipase at 37 °C. Data are shown as mean ± SD (n = 3).
Glutathione stability studies
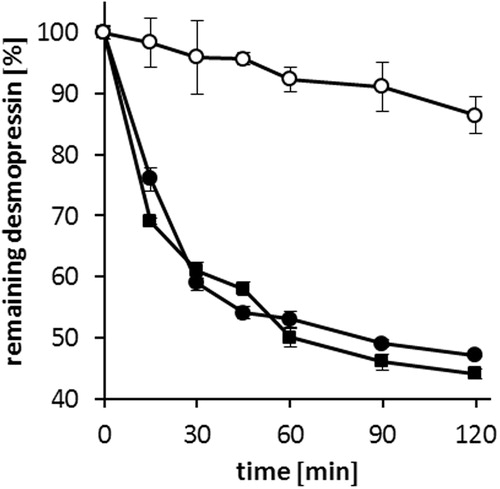
Biological thiols present in the gastrointestinal tract can originate from food (cysteine, homocysteine, N-acetyl cysteine, μ-glutamyl cysteine, glutathione) or endogenous reduced glutathione (Schmitz et al., Citation2006; Leonaviciute & Bernkop-Schnürch, Citation2015). The thiol-disulfide reactions in 50 mM acetate buffer pH 5.5 of desmopressin acetate, DES/AOT-SEDDS-F15 and DES/AOT-SEDDS-F4 with reduced glutathione (2.5 mg/mL) are presented in . Desmopressin acetate was degraded to 50% of its initial amount within 2 h, which correlates with already published results (Schmitz et al., Citation2006). Furthermore, no significant difference in degradation profile (p < 0.05) between desmopressin acetate and DES/AOT-SEDDS-F15 was observed. A rapid degradation of desmopressin in the first 15 min, where more than 20% of the initial amount was degraded, was measured. DES/AOT-SEDDS-F15 is composed of 80% hydrophilic surfactants. Formulations of this type are known to lose solvent capacity, which might be the case for diffusion of DES/AOT in the aqueous phase (Pouton, Citation2006; Pouton & Porter, Citation2008). In addition, a high amount of surfactants may also promote the diffusion of hydrophilic small molecule glutathione (307 Da) into droplets and its interaction with desmopressin. In contrast, a significant difference in desmopressin degradation profile (p < 0.05) was observed at DES/AOT-SEDDS-F4. After 120 min more than 90% of the initial desmopressin amount remained intact. Increased amount of water insoluble Capmul 907 P and lower amount of hydrophilic surfactants in DES/AOT-SEDDS-F4 compared with DES/AOT-SEDDS-F15 could have prevented the interaction between glutathione and desmopressin.
Figure 6. Degradation of desmopressin acetate (▪), DES/AOT-SEDDS-F15 (•) and DES/AOT-SEDDS-F4 (○) at 37 °C in 50 mM acetate buffer pH 5.5 by reduced glutathione. Data are shown as mean ± SD (n = 3).
Enzymatic stability studies
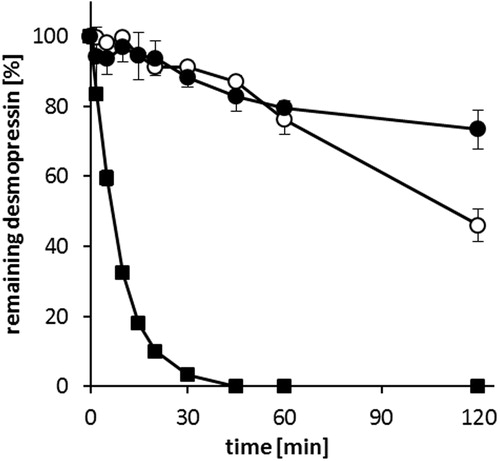
Desmopressin is relatively stable in the presence of trypsin and pepsin due to the removal of terminal amino group substitution of L-arginine by D-arginine (Matsui et al., Citation1985; Wang et al., Citation2002). However, it is readily degraded by α-chymotrypsin (Kahns et al., Citation1993; Fredholt et al., Citation1999; Christophersen et al., Citation2014). The degradation of desmopressin acetate, DES/AOT-SEDDS-F15 and DES/AOT-SEDDS-F4 are presented in . Desmopressin dissolved in phosphate buffer is inactivated within 45 min. In contrast, DES/AOT-SEDDS-F4 and DES/AOT-SEDDS-F15 have shown a protective effect toward α-chymotrypsin. After 60 min more than 80% of desmopressin in both SEDDS formulations remained intact. The protective effect indicates that the peptide remained incorporated in the oily droplets after emulsification of SEDDS in phosphate buffer (Hintzen et al., Citation2014; Karamanidou et al., Citation2015). In addition, certain excipients used in SEDDS formulations like Cremophor EL demonstrated to have enzyme inhibiting properties (Zhao et al., Citation1989).
Figure 7. Enzymatic degradation of desmopressin acetate (▪), DES/AOT-SEDDS-F15, (•) and DES/AOT-SEDDS-F4 (○) at 37 °C in 50 mM HEPES buffer pH 7.4 with 2 mM CaCl2 by α-chymotrypsin. Data are shown as mean ± SD (n = 3).
Resazurin assay
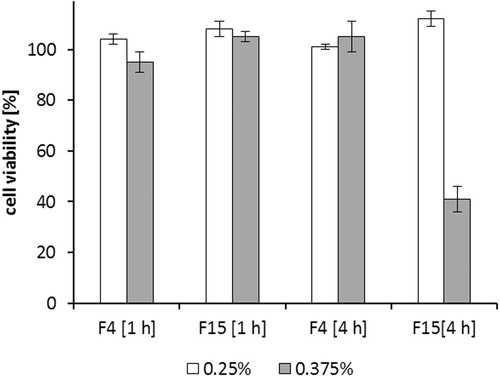
Usually the excipients with GRAS status, such as naturally derived oils and medium chain triglycerides are chosen for SEDDS development. However, many oils and surfactants may show cytotoxic effects if not used in low concentrations (Palamakula & Khan, Citation2004). To investigate cytotoxic effects of DES/AOT-SEDDS-F4 and DES/AOT-SEDDS-F15 a cell viability assay of a Caco-2 monolayer was determined. illustrates cytotoxicity of SEDDS after 4 h, which is considered to be in the range of an average small intestinal passage time. When applied in 0.25% (w/v) both SEEDS were nontoxic after 4 h. However, at increased concentration 0.375% (w/v) cell viability of DES/AOT-SEDDS-F15 was just 41% after 4 h. In contrast, DES/AOT-SEDDS-F4 in the same concentration appeared to be nontoxic. Surfactants are known to be harmful in high concentrations by interacting with cell membranes. DES/AOT-SEDDS-F15 is composed of 80% surfactants in comparison to DES/AOT-SEDDS-F4 containing only 40% surfactants likely explaining this observation. Additionally, Capmul 907 P can also act as a secondary surfactant and contribute to overall toxic effects.
Figure 8. Cell viability rates of Caco-2 cells after incubation for 1 h and 4 h with 0.25% (w/v) and 0.375% (w/v) DES/AOT-SEDDS-F4 and DES/AOT-SEDDS-F15. Data are shown as mean ± SD (n = 3).
Conclusion
Within this study, desmopressin was formulated in SEDDS using desmopressin-surfactant complexes. Two SEDDS formulations, DES/AOT-SEDDS-F4 composed of 40% hydrophilic surfactants and comparatively more hydrophilic DES/AOT-SEDDS-F15 containing 80% surfactants were investigated. DES/AOT-SEDDS-F4 demonstrated for the first time to our knowledge that SEDDS were able to protect the incorporated peptide against thiol–disulfide exchange reactions. It thus opens the door and gives new possibilities to design more efficient oral delivery systems for drugs containing thiol groups or disulfide bonds.
Acknowledgements
The authors would like to thank Michaela Hoffmann from Barentz Gmbh for Abitec excipients supply and technical support.
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
References
- Bandivadekar M, Pancholi S, Kaul-Ghanekar R, et al. (2013). Single non-ionic surfactant based self-nanoemulsifying drug delivery systems: formulation, characterization, cytotoxicity and permeability enhancement study. Drug Dev Ind Pharm 39:696–703
- Belhadj Z, Zhang S, Zhang W, Junlin W. (2013). Formulation development and bioavailability evaluation of a Self-nanoemulsifying Drug Delivery System (SNEDDS) of atorvastatin calcium. Int J Pharm 29:2051–5782
- Choi SH, Park TG. (2000). Hydrophobic ion pair formation between leuprolide and sodium oleate for sustained release from biodegradable polymeric microspheres. Int J Pharm 203:193–202
- Christiansen ML, Holm R, Kristensen J, et al. (2014). Cinnarizine food-effects in beagle dogs can be avoided by administration in a Self Nano Emulsifying Drug Delivery System (SNEDDS). Eur J Pharm Sci 57:164–72
- Christophersen PC, Zhang L, Müllertz A, et al. (2014). Solid lipid particles for oral delivery of peptide and protein drugs II–the digestion of trilaurin protects desmopressin from proteolytic degradation. Pharm Res 31:2420–8
- Fatouros DG, Deen GR, Arleth L, et al. (2007). Structural development of self nano emulsifying drug delivery systems (SNEDDS) during in vitro lipid digestion monitored by small-angle X-ray scattering. Pharm Res 24:1844–53
- Fosgerau K, Hoffmann T. (2015). Peptide therapeutics: current status and future directions. Drug Discov Today 20:122–8
- Fredholt K, Østergaard J, Savolainen J, Friis GJ. (1999). α-Chymotrypsin-catalyzed degradation of desmopressin (dDAVP): influence of pH, concentration and various cyclodextrins. Int J Pharm 178:223–9
- Friedl H, Dünnhaupt S, Hintzen F, et al. (2013). Development and evaluation of a novel mucus diffusion test system approved by self‐nanoemulsifying drug delivery systems. J Pharm Sci 102:4406–13
- Hintzen F, Perera G, Hauptstein S, et al. (2014). In vivo evaluation of an oral self-microemulsifying drug delivery system (SMEDDS) for leuprorelin. Int J Pharm 472:20–6
- Ilan E, Amselem S, Weisspapir M, et al. (1996). Improved oral delivery of desmopressin via a novel vehicle: mucoadhesive submicron emulsion. Pharm Res 13:1083–7
- Jannin V, Chevrier S, Michenaud M, et al. (2015). Development of self emulsifying lipid formulations of BCS class II drugs with low to medium lipophilicity. Int J Pharm 495:385–92
- Kahns AH, Buur A, Bundgaard H. (1993). Prodrugs of peptides. 18. Synthesis and evaluation of various esters of desmopressin (dDAVP). Pharm Res 10:68–74
- Karamanidou T, Karidi K, Bourganis V, et al. (2015). Effective incorporation of insulin in mucus permeating self-nanoemulsifying drug delivery systems. Eur J Pharm Biopharm 97:223--9
- Leonaviciute G, Bernkop-Schnürch A. (2015). Self-emulsifying drug delivery systems in oral (poly) peptide drug delivery. Expert Opin Drug Deliv 12:1703–16
- Mahjub R, Dorkoosh FA, Rafiee-Tehrani M, Bernkop Schnürch A. (2015). Oral self-nanoemulsifying peptide drug delivery systems: impact of lipase on drug release. J Microencapsul 32:401–7
- Matsui K, Kimura T, Ota K, et al. (1985). Resistance of 1-deamino-(8-D-arginine)-vasopressin to in vitro degradation as compared with arginine vasopressin. Endocrinol Jpn 32:547–57
- Meyer JD, Manning MC. (1998). Hydrophobic ion pairing: altering the solubility properties of biomolecules. Pharm Res 15:188–93
- Müllertz A, Ogbonna A, Ren S, Rades T. (2010). New perspectives on lipid and surfactant based drug delivery systems for oral delivery of poorly soluble drugs. J Pharm Pharmacol 62:1622–36
- Palamakula A, Khan MA. (2004). Evaluation of cytotoxicity of oils used in coenzyme Q10 Self-emulsifying Drug Delivery Systems (SEDDS). Int J Pharm 273:63–73
- Partenhauser A, Laffleur F, Rohrer J, Bernkop-Schnürch A. (2015). Thiolated silicone oil: synthesis, gelling and mucoadhesive properties. Acta Biomater 16:169–77
- Pouton CW. (2006). Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci 29:278–87
- Pouton CW, Porter CJH. (2008). Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev 60:625–37
- Rao SVR, Shao J. (2008). Self-nanoemulsifying drug delivery systems (SNEDDS) for oral delivery of protein drugs: I. Formulation development. Int J Pharm 362:2–9
- Schmitz T, Huck CW, Bernkop-Schnürch A. (2006). Characterisation of the thiol-disulphide chemistry of desmopressin by LC, mu-LC, LC-ESI-MS and Maldi-Tof. Amino Acids 30:35–42
- Shen H, Zhong M. (2006). Preparation and evaluation of self-microemulsifying drug delivery systems (SMEDDS) containing atorvastatin. J Pharm Pharmacol 58:1183–91
- Shen WC. (2003). Oral peptide and protein delivery: unfulfilled promises? Drug Discov Today 8:607–8
- Thomas N, Müllertz A, Graf A, Rades T. (2012). Influence of lipid composition and drug load on the in vitro performance of self‐nanoemulsifying drug delivery systems. J Pharm Sci 101:1721–31
- Wang J, Shen D, Shen WC. (1999). Preparation, purification, and characterization of a reversibly lipidized desmopressin with potentiated anti-diuretic activity. Pharm Res 16:1674–9
- Wang J, Wu D, Shen WC. (2002). Structure-activity relationship of reversibly lipidized peptides: studies of fatty acid-desmopressin conjugates. Pharm Res 19:609–14
- Wang J, Yadav V, Smart AL, et al. (2015). Toward oral delivery of biopharmaceuticals: an assessment of the gastrointestinal stability of 17 peptide drugs. Mol Pharm 12:966–73
- Zhao FK, Chuang LF, Israel M, Chuang RY. (1989). Cremophor EL, a widely used parenteral vehicle, is a potent inhibitor of protein kinase C. Biochem Biophys Res Commun 159:1359–67
- Zupančič O, Partenhauser A, Lam HT, et al. (2016). Development and in vitro characterisation of an oral self-emulsifying delivery system for daptomycin. Eur J Pharm Sci 81:129--36