
Abstract
Efficient drug delivery at vaginal cavity is often a challenge owing to its peculiar physiological variations including vast differences in pH. Keeping in view this attribute of the target site, the current work was aimed at developing formulation strategies which could overcome this and successfully deliver molecules like itraconazole through SLNs. Optimized SLNs with the given composition was selected for further development into mucoadhesive and thermosensitive gel. Stearic acid and Compritol 888 (1:1, w/w ratio) as lipid, a mixture of 3% Poloxomer 188 and 0.5% sodium taurocholate as surfactant and organic to aqueous ratio of 10:50 was taken. Carbopol 934 and Pluronic F 127 were taken for the development of gel. Optimized gel exhibited a desired gelling temperature (35 °C); viscosity (0.920 PaS) and appreciable in vitro drug release (62.2% in 20 h). MTT assay did not show any cytotoxic effect of the gel. When evaluated in vivo, it did not exhibit any irritation potential despite appreciable bioadhesion. A remarkable decrease in CFUs was also observed in comparison with control and marketed formulation when evaluated in rat infection model. Thus, the proposed study defines the challenges for developing a suitable formulation system overcoming the delivery barriers of the vaginal site.
Introduction
In the present research, we have tried to develop a model formulation with desired properties for site-specific vaginal delivery. Formulation attributes generally envisaged in a novel formulation include maximum drug loading, enhanced retention at the targeted site, and a sustained drug release throughout a therapeutic window. Vaginal cavity, as a delivery site, has often presented challenges of inter-individual variability owing to its wide range of physiological characteristics like pH variation, micro flora, cervical mucus, and cyclic changes during menstruation. The present work aimed at exploiting these physiological impediments for an effective site-specific drug delivery through a specifically designed formulation model. Vaginal candidiasis, a commonly reported affliction in women throughout the world, was selected as a target disease while itraconazole, a potent synthetic triazole antifungal drug with activities against broad spectrum of fungal species (Saag & Dismukes, Citation1988) was chosen as target drug moiety. Itraconazole has gained popularity in the literature as “practically impossible to apply to the body” (Barrett et al., Citation2008). Solid lipid nanoparticles (SLNs) have gained popularity for being a suitable carrier for hydrophobic drugs and have been successfully used for dermal and mucosal delivery. SLNs provide distinct advantages like good biocompatibility, easy to scale up and sterilize, control and/or target drug release, higher drug content, etc. Moreover, a combinatorial use of thermosensitive and mucoadhesive gel system would offer advantages like longer retention time and appropriate loco-regional outcomes with better control of infection.
Considering the peculiarities of the target site, drug and the disease, final dosage form comprising of SLNs loaded in a thermosensitive and mucoadhesive gel was developed, characterized, and evaluated.
Materials and method
Itraconazole was obtained ex gratia from Jubilant Organosys, Noida, India. Lipidic excipients used in the study were obtained as gift samples from Gattefosse India, except Cetyl palmitate (from Pioneer Organics, India) and stearic acid (from local market). Poloxamer was obtained as a gift sample from BASF (Germany). Sodium taurocholate was purchased from (Thomas Baker Ltd., Mumbai, India). All the excipients used have been approved for vaginal formulations by different regulatory agencies.
Formulation strategies for development of vaginal drug delivery system (VDDS)
The foremost step towards the development of a successful novel VDDS is to cogitate over physiological, anatomical, and microbiological aspects of the tract which could act as impediments for effective drug delivery. These should be explored well and exploited positively to ensure successful circumvention of the disease. The vaginal epithelium is covered with a thin film of fluid which is a mixture of several components like cervical mucus, secretions of Skene’s and Bartholin’s gland, transudates through the epithelium, leucocytes, endometrial, and tubular fluids (Paavonen, Citation1983). Women of reproductive age produce fluid at a rate of 3–4 g/4 h, while the discharge produced by postmenopausal women is reduced by 50% (Bergh, Citation1988). This mucus covering plays important role in drug absorption or action (Khanvilkar et al., Citation2001). Permeation through mucus is more complex than simple diffusion and includes additional effects like hydrodynamics and electrostatic phenomena. In classical (Fickian) description of mass transport in homogenous and dilute solutions, the diffusivity relates mass flux to concentration gradient. But in case of multi-component, heterogeneous system like vaginal mucus different strategies exist. The concentration of a particular chemical or biological moiety within mucus depends upon the permeability within mucus as well as resistance at the carrier–mucus interfaces. The carrier in this case may be a fluid or a polymeric device or film. Mathematical model of transport introduces an equilibrium partition coefficient (K), relating the equilibrium concentration of a solute in the two medium. In addition, non-equilibrium, transient behavior of partitioning may dominate during transport processes. As a drug must be in solution before absorption, the presence of a film or solution may be advantageous but a thick consistency may act as a barrier. But for localized drug delivery, it may act as a depot for a shorter period of time. The transport of drug that is poorly water soluble may be increased when the fluid volume is higher. In addition, thick vaginal mucus may assist in the bioadhesion of particles. A large volume of fluid generally sweeps out the drug or entire delivery system. Hence as per the author’s view, if a dosage is applied during the proliferative phase of menstrual cycle (lesser viscosity and comparatively higher cervical fluid volume), more positive results may be obtained.
The normal pH of the vagina in pre-menopausal women ranges from 4.5 to 5.5 which rises with an increasing amount of cervical mucus, progression of menstrual cycle, and coitus. It is maintained by lactobacilli that convert glycogen from exfoliated epithelial cells into lactic acid, playing an important role in the control of infection by common pathogens (Richardson & Illum, Citation1992). In most of the women, pH gradient exists; pH is lowest near the anterior fornix and highest near the cervix. Since most of the drugs are weak electrolytes, the pH may change their degree of ionization and may also affect the release profile from VDDS. Using the Henderson–Hasselbalch equation, we may say that when the pKa of the acidic drug is ≥6, absorption is more in normal vaginal milieu while drugs having pKa ≤ 5 remain ionized and localized. Opposite is the condition with weakly basic drugs. So, accordingly we may sort out molecules for local and systemic action. Another characteristic of the molecule to be considered is the log P, whose value should be > 2 to accumulate subcutaneously or intramuscularly. Otherwise molecules having log P ≤ 2 offer themselves as potential candidates for transdermal delivery.
Vaginal epithelial cell layer, which is about 200–300 μm thick, is an elaborate system of channels capable of changing width in certain cell layers with the menstrual cycle (Song et al., Citation2004). Like other mucosal delivery routes, drug transport across the vaginal membrane may occur by a number of different mechanisms namely (a) diffusion through cell according to concentration gradient (transcellular), (b) receptor-mediated transport, and (c) diffusion through tight junction of cells (intercellular) (Hwang et al., Citation1977). When we talk of the size of the molecule, animal models like monkey and rabbit have shown a continuous increase in the absorption with the increase of chain length (Benziger & Edelson, Citation1983). On the contrary, surface-active agents which give promising results in nasal and rectal absorption of drugs have insignificant effect in case of vagina.
On one hand, the molecular characteristics required for materials to be bioadhesive have been recognized and can be listed as the presence of hydrogen bonding groups (OH, COOH), strong anionic charges, flexibility to allow penetration in the mucus network, surface tension properties appropriate for wetting the mucus or mucosal tissue surfaces, a high molecular weight and fast hydration capability, as this will help in the formation of polymer interactions and entanglements necessary for adhesion. On the other hand, interaction between nanostructured lipidic carrier and subcutaneous lipid has already been hypothesized (Ricci et al., Citation2005). In fact, both lipidic and polymeric nanoparticles have shown the peculiarity to reduce the permeation of the drug molecule through the skin while enhancing the penetration into the upper skin layers (Jenning et al., Citation2000). On the contrary, the lipid bioadhesive properties have to be accounted as they may play a relevant role.
Selection of drug candidate
Molecular weight cut off for drug absorption in case of vaginal membrane is higher than any other mucosal surface. Thus if we take a molecule having comparatively larger size, low aqueous solubility, released in a sustained manner and showing better interaction with bovine serum albumin (BSA), we could expect the molecule to remain in vaginal milieu for a longer time rather being absorbed systemically or leaked out of the cavity. BSA (Sigma-Aldrich, Bengaluru, India) was taken as a model protein to see the interaction as it is the only proteinous constituent of simulated vaginal fluid (Owen & Katz, Citation1999). Azole class of compounds (Itraconazole, Fluconazole, Ketoconazole, and Micoconazole) was chosen for the study, considering their antifungal spectrum, molecular size, and low water solubility. All the compounds were obtained as gift samples from pharmaceutical companies (Jubilant Organosys, Mumbai, India and Ranbaxy Laboratories Limited, Mumbai, India).
The Autodock Vina 1.1.2 (Trott & Olson, Citation2010) was employed to study the interaction between Azole compounds and BSA. Minimized energy levels of ligands were generated by MM2 module of ChemBio3D 12.0 (ChemDraw, San Diego, CA). Energy of four ligands was minimized to RMS gradient value 0.01 before docking. 3D structures were visualized by PyMOL. Rigid docking study was performed using the available X-ray crystal structure of Bovine serum albumin. (PDB ID: 3V03).
Selection of lipid
On the basis of interaction with Bovine serum albumin
The general criterion of lipid selection for the development of SLN is drug solubility in a particular lipid. On one hand, to obtain a better biodistribution, stabilizers (surfactants) are selected after studying interaction with proteins (Goppert & Muller, Citation2005) which results into maximal in vivo residence time. But stabilizers, if not bounded chemically, are loosely attached to the colloidal surfaces. Also, their micellar network is concentration dependent and it is expected that after sufficient dilution they may leave the surface. On the other hand, chemical and enzyme-catalyzed processes are the main forces which may lead to degradation of stabilizers. The enzymatic degradation of such polymers depends on a number of factors (apart from those related to the polymer chemical structure) like, temperature, pH, electrolyte concentration, presence of enzyme inhibitors, presence of other organic compounds (e.g., urea), cations (certain systems), and other co-solutes. Thus, when the stearic hindrance by the stabilizers is minimized around colloidal particles (e.g., solid lipid nano particles), it is the core of particle (lipid in case of SLN) which interacts with body fluids. So, an attempt has been made here to study the preferential adsorption of a protein over different solid lipids available. BSA has been chosen for the current study, considering the fact that it is the only proteinous constituent of simulated vaginal fluid (SVF) (Owen & Katz, Citation1999) which is used widely in several in vitro studies and is a much studied protein. Hypothesizing that once a lipid is rendered without the coatings of stabilizers, it is its inherent affinity to remain adhered to the body fluids/proteins that will allow it to remain dispersed within the body. Otherwise it may precipitate out and show poor retention. For the above hypothesis, following experimental set up was run.
About 1.5 g each of selected lipids (lipids showing better drug solubility) were passed through sieve number 80 and stored in desiccators. A 15 ml glass syringe (inverted) was taken and its nozzle blocked with a filter paper. Weighed quantities of lipids were then filled into this assembly and a 10 ml of freshly prepared BSA solution (2 mg/ml) was added into it. The assembly was then left undisturbed at room temperature (25 °C) for an hour. Thereafter, the cap was dismantled and the samples were collected and analyzed by UV spectrophotometer (Shimadzu, UV 1601, Shimadzu Corp., Tokyo, Japan) at 279 nm. Calibration curve of BSA was plotted and used for the calculations (y = 0.1265x − 0.0132; r2 = 0.996).
On the basis of maximum crystal lattice space (d value)
After sorting out the lipids on the basis of preferential adsorption with bovine serum albumin, we developed combinations of them to generate maximum lattice space within the bulk. All the lipids, either single or in mixture (1:1, w/w ratio), were dissolved in chloroform and then were dried slowly. They were pulverized to get uniform mass size. X-ray diffraction (PW 1830, Phillips, Bangalore, Karnataka, India) patterns of all the samples were recorded between 2θ = 8° and 88° at 35 kV and 30 mA, respectively.
Selection of surfactant
A high surfactant concentration favors a smaller particle size, a narrower particle size distribution, and long-term stability of lipid nanoparticles, but it is generally believed that increasing the surfactant concentration beyond a level increases the toxicological potential irrespective of the site. Hence, a balance needs to be maintained. For the selection of surfactant (or combination), placebo SLNs were formulated and percentage transmittance was determined. % Transmittance was also determined after 3 d to see the effect of stabilizers on particle growth (size ripening).
Development of SLN with solvent emulsification/evaporation method
The solid lipids (Stearic acid, Compritol 888, Compritol E ATO, and their combinations) were dissolved in small quantity of dichloromethane; bath sonicated for 2 min and total volume was made up to 10 ml. It was considered as an organic phase. Aqueous phase was made in such a manner that after the addition of organic phase, total concentration of surfactant follows the values shown in . Sodium taurocholate (0.5% w/v) was taken as a co-surfactant to generate sufficient negative charge over the SLNs. Poloxamer 188 was taken as a stearic surfactant in a variable quantity (2–5% w/v). Organic phase was then added drop wise (with the help of syringe) into aqueous surfactant solutions and simultaneously it was given high energy with the help of probe sonicator for 3 min with a pause of 10 s after every minute.
Table 1. Trails of SLN optimization and their evaluation (% entrapment efficiency, % drug content, mean particle size, Zeta potential, and particle distribution index, PDI).
The colloidal suspension was then filtered through Whatman filter paper (Jay Scientific Company, Mumbai, India) and left for stirring (1500 rpm) for 4 h to remove the chloroform. The suspension was frozen for 24 h in a Lyph-lock apparatus and then freeze dried (Dry winner, DW-8-85 Heto Holten, Denmark) for 12 h. Mannitol (USP grade; Sigma-Aldrich, Bengaluru, India) (1% w/v) was added as a cryoprotectant. A batch of optimized formulation (T 10) without cryoprotectant was also freeze dried for comparative characterizations.
There was a reason to adopt this method for SLN production and avoiding well-established methods like high-pressure homogenization, hot homogenization, and others. Increased temperature and use of higher amount of surfactant allows partitioning of drug into water phase and subsequent re-partitioning to the organic phase, consequently the drug concentrates in the outer shell and/or on the surface of the particle (Goppert & Muller, Citation2005). The molecules that do not accumulate in the core are responsible for the phenomena of “burst release”. Hence to have a more sustained action of the final formulation, we adopted above-mentioned method.
Characterization of SLNs
Following characterizations were performed for the developed SLNs.
Particle size and zeta potential (ξ) analysis
Particle size was determined using a photon correlation spectrometer (PCS; Zetasizer-1000 HAS, Malvern Instruments, Malvern, UK) based on the laser light scattering phenomenon. Freeze dried powders were dispersed in deionized water at 25 °C under gentle shaking and filtered through 0.22 μm membrane filter (Pall India Pvt. Ltd, Hinjewadi, India) in order to eliminate multi-scattering phenomenon and experimental errors. Average particle diameter (Δdm), polydispersity index (p¡), and zeta potentials (ζ) were recorded. All measurements were done in triplicate using disposable polystyrene cuvettes (Malvern Instruments, Malvern, UK).
Differential scanning calorimetry
In order to characterize the thermal behavior of the lipid nanocarrier and bulk lipids, DSC was performed using a heat influx instrument (DSC-7, Perkin Elmer Pyris 6 Instrument, Norwalk, CT) according to method described earlier (Mirza et al., Citation2012).
Powder X-ray diffraction
X-ray diffraction study of SLN was performed according to the method described in the previous section.
Surface morphology studies
Surface morphology of SLN was studied with the help of transmission electron microscope (TEM) (TOPCON 002B) operating at 200 KV, capable of point-to-point resolution. Properly diluted samples (1/100 in water) were used for TEM observations. A drop of diluted suspension of SLN was allowed to deposit directly on the circular copper film grid and observed after drying.
% Entrapment efficiency (% EE)
The % EE of itraconazole into SLN was determined by the ultrafiltration method using centrifugal filter tubes with a molecular weight cutoff of 30 kDa (Amicon Ultra, Millipore, Ireland). The concentration of itraconazole in SLN and in the ultra-filtrate (free drug) was analyzed by a slight modification of a validated HPLC method (Yoo et al., Citation2000), with an accuracy of 95.5%, a precision of 6.7%, a regression coefficient of 0.998, a limit of detection 5.6 ng/ml, and a limit of quantification of 16.4 ng/ml. The instrumental parameters were according to our previous reports (Mirza et al., Citation2012). Rt of itraconazole was observed at 8.25 min. The % EE was calculated using the following equation:
Possible lipid interferences with itraconazole were also investigated by comparing the standard curves of drug alone and in the presence of lipids. The differences observed between the standard curves were within the experimental error, thus inferring that no lipid interference occurred.
Determination of residual solvent content
The residual solvent content in samples was quantified by GC-MS system (Agilent 7890A series (Waldbronn, Germany)) equipped with a split–split less injector and CTC-PAL auto sampler (Mirza et al., Citation2012).
Lipid stability by GC-MS study
For a successful and stable formulation of SLN, the chemical stability of excipients used for particle production is a prerequisite. Chemical analysis of lipids was performed by gas chromatography using a GC-MS system (Agilent 7890A series (Waldbronn, Germany)) equipped with a split–split less injector and CTC-PAL auto sampler as described previously. Determination was performed via the corresponding methyl esters of the fatty acids of the lipids. Transmethylation of the lipids was done according to the method described (Garces & Mancha, Citation1993). In order to separate the lipid from the SLN dispersions, the formulations were centrifuged at 17 000 rotations/min for 30 min. About 50 mg of the obtained lipid phase were mixed with methylating mixture containing methanol/toluene/dimethoxypropan/H2SO4 (39:20:5:2, by volume) in tubes with teflon caps. The quantity of methylating liquid was 3.3 ml, and Heptan was added to a total volume of 5 ml. This mixture was incubated in a water bath at 80 °C for 90 min. The tubes were cooled to room temperature and shaken, two phases were formed. The upper phase contained the fatty acid methyl esters. The carrier gas was helium at the flow rate of 1.5 ml/min.
Development of mucoadhesive and thermo sensitive gel incorporating SLN
Among the various vaginal dosage forms available, patients are known to tolerate gels better than inserts or ointments (Edsman et al., Citation1998). In order to substantially enhance muco-retention and obtain a sustained drug-release profile, SLNs were further incorporated into gels. Carbopols are known to have mucoadhesive properties and have been used in the formulation of vaginal delivery systems (Wang & Lee, Citation2002; Baloglu et al., Citation2003). Carbopol (CP 934) is also reported to have the highest bioadhesion potential at vaginal pH (Repka & McGinity, Citation2001). Poloxamer 407 (P 407) or Pluronic F127 has been preferably used as a thermo-sensitive polymer which shows excellent water solubility, good drug release characteristics, low toxicity, and irritation, and has compatibility with other excipients (Zaki et al., Citation2007).
Thermosensitive and mucoadhesive gel formulations were prepared according to the cold method (Choi et al., Citation1998). Mucoadhesive polymer, Carbopol 934 (Sigma-Aldrich, Bengaluru, India) (0.2% w/v) was slowly added to citrate phosphate buffer (0.1 M, pH 4.0) at 4° C with gentle mixing for 3 h and allowed to hydrate. Then it was centrifuged at 3000 rpm for 15 min to remove the air bubbles. Care was taken to allow minimum air bubbles level during handling. Pluronic® F 127 (15%, 18%, and 20%, w/v) was then added to CP 934 solution and allowed to dissolve overnight at 4 °C. Optimized SLN (T 10) was dispersed in Pluronic solution in such a way that the total strength of itraconazole in gel remained 1% w/v. Developed gels were studied for following parameters.
Measurement of sol-gel transition temperature (Tsol/gel) and gelation time
Five milliliter of the gel was placed in a 20 ml (internal diameter ∼30.7 mm) transparent vial containing a magnetic bar (dimension ∼15.0 × 8.2 mm) with a constant (150 rpm) stirring (2 MSH magnetic stirrer, HICON, Delhi, India). Vial was further placed in a water bath, which was heated (from 4 °C) at a rate of 2 °C/min. When the magnetic bar stopped moving due to gelation, the temperature displayed on the thermistor was determined as the gelation temperature.
Viscosity measurement
The viscosity of the gels was determined by using Brookfield R/S plus rheometer (Brookfield Engineering, Middleboro, MA) using a C50-1 spindle in triplicate mode. The temperature of the laboratory was maintained at 37 °C. Sufficient time was given to the formulation to reach at room temperature as it was stored at 4 °C.
In vitro drug-release kinetics
The volume of the vaginal fluid might be much smaller than the volume of buffer medium used in vitro. Moreover, the fixed stirring conditions used in vitro may not simulate the vaginal flux of fluids. Given these differences between in vitro test conditions and in vivo vaginal conditions, we cannot directly correlate the in vitro dissolution result to the in vivo situations. Dissolution studies were performed according to our earlier reported method (Chopra et al., Citation2007). Method includes USP XXVIII, paddle-rotating method (Electrolab dissolution tester, Electrolab, Mumbai, India) at 37 °C ± 0.5 °C and 75 rpm using phosphate buffer (pH 4.5) as a dissolution medium. The gels were first packed into a dialysis bag and placed into the dissolution media. Dissolution studies were carried out in triplicate, maintaining the sink conditions for all the formulations. A 5 ml aliquot of sample was withdrawn at regular time intervals, filtered, and assayed. The level of itraconazole in the media was estimated using the previously reported HPLC method (Yoo et al., Citation2000).
In vitro cell toxicity studies of gel (G4)
The cytotoxicity of gel was evaluated by 3-(4-5-dimethylthiazol-2-4)-2, 5-diphenyltetrazolium bromide (MTT) assay using HeLa-S3 cell lines. HeLa-S3 cell lines were obtained from National Center for Cell Science (NCCS, Pune, India) and the process was carried out by our previously reported method (Mirza et al., Citation2011).
Preclinical evaluation of gels
Bioadhesion and irritation tests
In vivo bioadhesion studies were carried out after taking permission from Institutional Animal Ethical Committee of Jamia Hamdard (proposal no. 712). Experimentation was carried out according to the earlier reported method (Chopra et al., Citation2007). Briefly, 0.5 g of the gel was mixed with 0.4% trypan blue dye. Resulting dark blue colored formulation was inserted into the rat vagina with a 2 ml plastic syringe (without needle). For each group, three rats were taken. After 24 h of administration, the rats were sacrificed and the retention of bioadhesive formulation at the administration site was visualized by dye intensity. For the control group, simulated vaginal fluid (pH 4.5) was taken.
Rats have simple, cuboidal, or columnar epithelium which is highly sensitive to mucosal irritation when compared with human vagina. Adult Wistar rats (3 months old; 150–180 g) were used for the in vivo toxicity studies. Tap water and food pellets were available ad libitum throughout the study. Room temperature was maintained at 25 ± 2 °C with a relative humidity 50 ± 5%. Three rats were taken to each group. The rats were treated once daily with 0.5 g of gel for 14 d. Prior to every administration, rats were evaluated for vaginal or vulval irritation, discharge, or bleeding from vagina. After the completion of 14 d study period, they were scored for possible erythema. The mean erythemal scores were recorded (ranging from 0 to 4) depending on the degree of erythema as follows: no erythema = 0, slight erythema (barely perceptible-light pink) = 1, moderate erythema (dark pink) = 2, moderate to severe erythema (light red) = 3, and severe erythema (extreme redness) = 4. Then, they were sacrificed and transverse section of the vaginal tissue was examined by a pathologist (Singh Histopathology Processing Center, New Delhi, India) for the severity of epithelial loss and atrophy. The control group was given the same volume of SVF.
In vivo antifungal studies
A rat infection model was used to study the final formulation in vivo (De Bernardis et al., Citation1997) after getting approval from Institutional animal ethical committee of Jamia Hamdard (proposal no. 742). Eighteen female Wistar rats were divided in groups of three for the application of optimized gel, Gynazole (2% w/w), and control (SVF treatment). Six days prior to inoculation of infection, all animals were maintained under pseudoestrus by subcutaneous administration of estradiol benzoate. Into these oophorectomized rats, 0.1 ml of C. albicans (108 cells per ml of saline) was inoculated. Thereafter, 1 g of G4 (1% w/w) and 0.5 g of Gynazole (2% w/w butoconazole nitrate) were applied. During specified time intervals, 1 μL of the fluid from vaginal cavity was withdrawn, diluted, and spread on Sabouraud agar (Himedia Laboratories, Mumbai, India) containing chloramphenicol (50 mg/ml). It was then incubated at 32 °C in the BOD incubator (LHC-78-Labhospmake, Karnataka, India) for 48 h and colony forming units (CFUs) were counted. A rat was considered infected when at least 1 CFU was present in the vaginal sample (i.e., a count of ≥103 CFU/ml). A graph was plotted between time (days) and number of CFUs present in the vaginal fluid.
Results and discussion
Selection of drug candidate
Results of the docking studies indicated that lowest energy level (−9.7 kcal/mol) was obtained with itraconazole which is an evidence of strongest interaction with protein BSA. Thus, itraconazole appears to be a suitable candidate. Clinical investigations has already indicated that application of 5 g of a 2% itraconazole cream was very well tolerated and the drug was not systemically absorbed (Francois et al., Citation2003). As per the previous discussion, it was also worthy to use itraconazole for a successful development of VDDS considering its pKa (5.6) and normal vaginal pH (4–5). The drug has an interesting tissue distribution, which has made possible effective and rapid treatments of vaginal candidiasis when the drug is administered orally (Bloch et al., Citation1988). A topical itraconazole containing formulation may be of use for several reasons including the opportunity to generate high local tissue levels, more rapid drug delivery, and lower systemic exposure. This may be especially important for treating pregnant patients.
Selection of lipid
Interaction with bovine serum albumin
Result obtained with the study is shown in . The total volume of sample recovered was less than the volume of BSA used. The amount of BSA adsorbed by 1.5 g of lipid was determined in the order of Compritol E ATO > Compritol 888 ATO > Stearic acid > Precirol Cetyl palmitate > Gelucire (39/01) > Gelucire (50/13). Thus, lipids selected for further studies were Compritol E ATO, Compritol 888 ATO, and Stearic acid.
Figure 1. (A) Preferential adsorption of bovine serum albumin with different lipids. Lipids from various sources and chemical compositions were taken for the study and evaluated comparatively. (B) X-ray powder diffractograms of single and binary mixtures of lipids. In the binary mixtures, 1:1 ratio of lipids was used. Patterns of all the spectra were found to be overlapping.
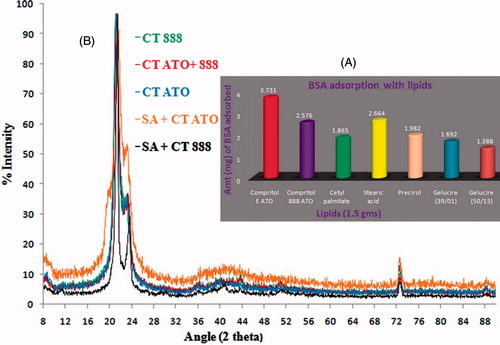
Preferential interaction of BSA with lipidic colloidal particles is well supported by the literature (Almeida & Souto, Citation2007). A double hydrogen-bonding mechanism for stearic acid binding to BSA has been described (Ge et al., Citation1990). The ability of the acid to form two hydrogen bonds apparently fixes it more rigidly in the protein, preventing rotation about either of the single-hydrogen bond. A double-hydrogen bonding mechanism is most consistent with the formation of a salt bridge between the negatively charged carboxylate of the acid and either a positively charged guanidino group of arginine or the positively charged omega-amino groups of two lysine residues. On the contrary, Compritol 888 ATO (glyceryl behenate) is chemically a mixture of mono-, di-, and triglycerides of behenic acid (C22). It is also expected to interact with BSA by the formation of some hydrogen bonds.
On the basis of maximum crystal lattice space (d value)
Slow evaporation of chloroform provided enough time to minimum energy and stable lattice arrangement. All the processed lipids showed maximum counts near 21° (Baloglu et al., Citation2003). On one hand, using Bragg” equation (nλ = 2d sin θ), d values for all the lipids were calculated at an angle which showed maximum counts (). XRD spectra obtained from the analysis are shown in . Thus, it is clear that the powders are not having a long range of crystalline pattern. In a solid lipidic carrier, it is expected that a solid with less orders in lattice could accommodate maximum amount of drug. On the other hand, maximum d value was obtained in the case of Compritol 888 + Compritol E ATO (4.3581) and Stearic acid + Compritol 888 (4.3502). Thus, we might expect that these combinations of lipids would accommodate larger amount of drugs.
Table 2. X-ray diffractogram interpretation of lipids and their physical mixtures (1:1 ratio).
Selection of surfactant
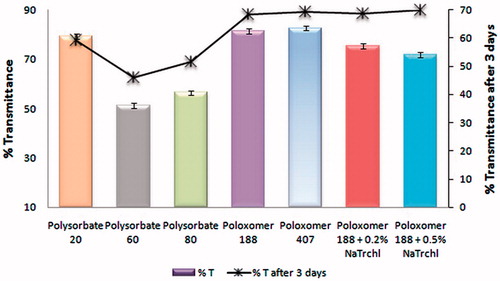
A SLN suspension with high % transmittance may be correlated with smaller particle size and maximum adsorption of surfactants over colloidal particles. Maximum transmittance was observed with Poloxamers, 82.6% with Poloxamer 407 and 81.3% Poloxamer 188. But for further studies, a Smix (a mixture of surfactants) of (5% P 188 + 0.5% Na Taurocholate) was chosen because the combination gave a negligible decrease in % transmittance after 3 d ().
Figure 2. Transmittance studies of nanosuspension using different stabilizer(s). After the first reading, the nanosuspensions were left undisturbed for 3 d to evaluate stability.
Characterization of SLN
SLN based on single lipids exhibited limited drug payloads and drug expulsion from the crystal lattice as the lipids have a tendency to re-crystallize in due course of time. During re-crystallization, the surface area of the particle would increase remarkably which would add to the instability to the colloidal system. Cryoprotectants are space holders which prevent the contact between discrete lipid nanoparticles. Furthermore, they interact with the polar head groups of the surfactants and serve as a kind of “pseudo hydration shell” (Mobley & Schreier, Citation1994).
Particle size and zeta potential (ξ) analysis
Obtained zeta potentials exhibit sufficient stability of the SLNs. Lipid particles are generally negatively charged (Schwarz & Mehnert, Citation1999) and colloidal system is found to be stable when ξ(>|30| mV) due to electrostatic repulsion (Levy et al., Citation1994). This is valid only in the case of sole electrostatic stabilization. Electrostatic stabilization and stearic stabilization effects are additive. Hence, for the development of SLN, we have taken a combination of surfactants (Poloxamer and sodium taurocholate). It has also been suggested that without stearic stabilization, a nano-particulate system would not be stable even if ξ value is greater than 30 mV (Tadros & Vincent, Citation1980). A certain thickness of polymer layer is necessary (>10 nm) for efficient and complete stearic stabilization (Washington, Citation1996). According to the theory of DLVO, a system could be regarded as stable if the electrostatic repulsion dominated the attractive Van der Waals forces. Non-ionic surfactants contribute to the stability by offering stearic stabilization, while ionic surfactants contribute by generation of electric double layer. Increasing the amount of stearic stabilizer (Poloxamer 188) resulted into higher zeta potential values (). Non-ionic surfactants could not ionize like ionic surfactants but demonstrated their role in zeta potential. The reason might be due to molecular polarization and the adsorption of emulsifier molecule on the charge in water, it was absorbed to the emulsifier layer of particle/water interface and electric double layer similar to ionic was found. Like in the case of P 188, less polar poly (propylene oxide) chain were dissolved and segregated into a hydrophobic micelle core surrounded by a soft “brush” of highly hydrated flexible poly (ethylene oxide) chain.
Average size of the particles and their size distribution are also given in . Mean particle size was bigger when stearic stabilizer was used alone. In the given set of experiments, we achieved a size range of 250.2–546.9 nm which is acceptable in the case of topical drug delivery. From the table, it is evident that with an increase in surfactant value, the size of the particle is falling sharply. The use of lipidic mixture also helped in decreasing the size of particles. The difference in polarity of the molecules (stearic acid versus Compritol) should have a minor influence on the particle size as the interfacial phenomena are supposed to be dominated by the properties of the surfactants.
Differential scanning calorimetry (DSC)
DSC thermo grams of bulk lipids (single or mixture) and drugs are given in and of different SLNs are given in . A sharp melting endotherm at 170.408 °C was observed for itraconazole. Melting enthalpy (ΔH), melting point, crystallinty index, and peak width of bulk materials and SLNs are given in .
Figure 3. Overlay of differential scanning thermograms (heat flow as a function of temperature) of itraconazole, Single lipids (stearic acid and compritol) and binary lipids. In all the samples, the processes were found to be endothermic in nature. Melting temperatures of all the lipid(s) were not too separated.
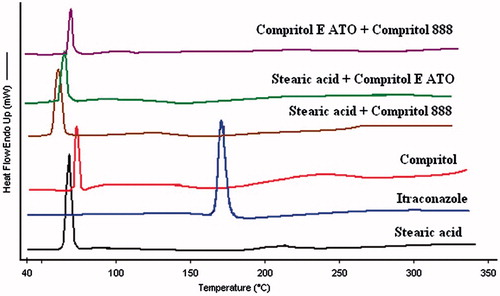
Figure 4. Overlay of differential scanning thermograms (heat flow as a function of temperature) of different solid lipid nanoparticles. In all the samples, the processes were found to be endothermic in nature and there were not any sharp peak rather broad humps were there. Also the melting temperatures of all the SLNs were not too separated.
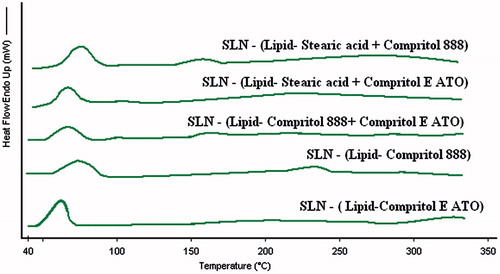
Table 3. Results of the DSC thermograms of single lipid, binary mixtures, and their SLNs.
It was evident from the thermo grams that itraconazole was in amorphous state whether it was present in core or outer shells of the particle. The amorphous form of a drug has a higher thermodynamic and chemical potential (Weuts et al., Citation2003), while the crystalline state has the lowest total energy content. The higher thermodynamic activity of the drug can produce supersaturated solutions (Betageri & Makarla, Citation1995) and can provide an opportunity to enhance solubility. One might also expect that the complete absence of long-range three-dimensional inter-molecular order associated with any material might significantly modify the mechanical and physical properties also (Hancock et al., Citation1999).
Organic substances usually show a melting range. An increased melting range could be correlated with impurities or less ordered crystals but the various instrumental analyses used in the current work have ensured lowest levels of impurities. A manual method of measuring melting range is peak width (i.e., difference between onset and maximum). Peak width of SLNs was found to be higher (two or three times) than any of the bulk material (itraconazole of bulk lipids). Melting enthalpy (ΔH) is another characteristic of the crystal order if the influence of impurities is ignored. For the less ordered crystal or amorphous state, the melt of the substance does not require or just requires less energy than the perfect crystalline substance which is needed to overcome the lattice force. As a result, the higher melting enthalpy values should suggest higher ordered lattice arrangement and vice versa. In this perspective also, the ΔH value of SLNs was smaller than the bulk materials from which they have been developed.
Another phenomenon we could observe from the thermos grams is the reduction of melting temperature of colloidal lipid particles as compared with bulk material which has been attributed inter-alia to their small size and the presence of surfactants. It can be discerned by the Gibbs–Thomsan equation which in itself is derived form of the Kelvin equation. It reflects the decrease in melting temperature for a particle of given size compared with the bulk material and becomes particularly more pronounced in the lower nanometer size range:
where T is the melting temperature of a particle with radius r, T0 is the melting temperature of the bulk material at the same external pressure, γsl is the interfacial tension at the solid–liquid interface, VS is the specific volume of the solid, ΔHfus is the specific heat of fusion.
Broadening of peak possibly results due to the presence of particle fractions with variable size which melt at different temperatures. The loading of the drug did not provoke any considerable effect in the lipid matrix thermal behavior under these experimental conditions.
In order to compare crystallinity between the developed formulations, a useful parameter is % crystallinity index which is defined as the percentage of the lipid matrix that has re-crystallized during storage time. Following equation is used to calculate % CI:
Thus, we see that highest crystallinity was developed by SLN (stearic acid + Compritol E ATO) with respect to its bulk lipid while the lowest crystallinity was developed by SLN (Compritol 888) with respect to its bulk lipid.
Powder X ray diffraction
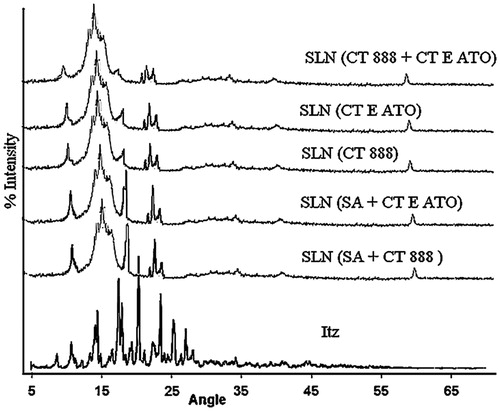
A diffractogram (2θ versus % intensity) can be considered a fingerprint of the crystal structure and provides a commentary with different crystallographic status of the system. One peak will be exhibited for all repeating planes with the same spacing. Itraconazole powder was crystalline in nature () and showed characteristic peaks at 14.1°, 17.8°, 20.6°, 23.2°, 25.7°, and 26.5°. Overlapping images of different bulk lipids are shown in . Prepared SLNs were not having characteristic peaks of itraconazole except some diminished peaks around 21° and 23°. Thus, corroborating the results with DSC, we may say that itraconazole is present in the amorphous state in SLN. The production of nanoparticles also reduces the degree of crystallinity. The reduction in crystallinity may be due to the partial formation of lower energy lipid modifications. In addition, surfactants distributed to the melted lipid phase during the production process can also distort crystallization. Broad hump around 19° is present in all the SLNs which may be the shifted peak of lipids as seen in . Thus, we see that lipids in all the cases have attained their small-ranged lattice pattern. Minutely considering the diffractogram, all five images of SLN were divided into three groups. Diffractogram of SLN of SA + CT 888 was matching with SA + CT E ATO, diffractogram of CT 888 was similar with CT E ATO, while diffractogram of CT 888 + CT E ATO was distinct from others. Thus, we can deduce that with different compositions of lipids, different lattice patterns are developed.
Figure 5. Overlay of X-ray diffractograms of itraconazole and different SLNs. The angles were kept wide. The patterns clearly indicate the change in crystallinity patterns.
Surface morphology studies
The shape of the lipidic nanoparticles is appearing round and homogenous. Particle size in the image is ranging from 196 to 395 nm (Supplementary Figure 1). More precise information on particle size and size distribution has been described early.
% EE
The outcome of % EE () was in accordance with the results obtained from XRD and DSC studies, i.e., as we break the crystalline lattice of solid by developing a mixture, we could accommodate more amount of drug. Highest d values were noted with mixtures of SA: CT E ATO and SA: CT 888 which translated into maximum entrapment of drug in the lattice. Enthalpy (ΔH), which is an indirect estimation of crystallinity of the material, was the lowest in the case of these two mixtures. Thus, we may devise that by the chosen method, we may lead to substantial imperfections in a carrier resulting into higher drug loading.
Considering the different parameters T 10 was selected as an optimized SLN.
Determination of residual solvent content
Dichloromethane was found to be absent in all the SLNs. Recommended limit of ICH Q3C guideline (ICH, 1997) in the case of dichloromethane is 6 mg/d and the residual amount limit in USP XXIII is 6 mg/d.
Lipid stability by GC-MS study
After keeping the SLN at room temperature for 3 months, lipid stability was performed. Data obtained are given in . There is no major decomposition in any case. It may be due to the fact that the surfactant concentration was low and the incubation time was small. In another study, we have also kept batches for a long-term study. These batches are loaded comparatively with higher surfactant concentration.
Characterization of gel
Measurement of sol-gel transition temperature (Tsol–gel), gelation time, and viscosity
The gelation temperature is defined as the point where the elasticity modulus is half way between the values for the solution and for the gel. Carbopol has been reported to interact strongly with low molecular weight drugs and cause precipitation (Blanco-Fuente et al., Citation2002). But in our case, physicochemical status of drug or SLN does not affect gelling temperature significantly. Different gelation temperatures are given in . With an increasing fraction of P 407, there was a decrease in gelation temperature. Gelation could happen at as low as 4 °C if polymer concentration is kept constant. Gelation phenomenon is a result of body-centered cubic packing of spherical micelles. Temperature plays an important role in the micelle formation through temperature-dependent hydration of the ethylene oxide units. Water is a good solvent for poly (ethylene) oxide –PEO – as well as poly (propylene) oxide – PPO chains of polymer at low temperatures. However, at higher temperature, the solubility of PPO is reduced and micelle formation occurs. At low temperatures in aqueous solutions, a hydration layer surrounds P 407 molecules. However, when the temperature is raised, the hydrophilic chains of the copolymer become desolvated as a result of the breakage of the hydrogen bonds that have been established between the solvent and these chains. This phenomenon favors hydrophobic interactions among the polyoxypropylene domains, and leads to the formation of gel. Aqueous solutions of poloxamers are stable in the presence of acids, alkalis, and ions (Escobar-Chavez et al., Citation2006), so they may also be expected to form gel in vaginal milieu.
Table 4. Gelation temperature and viscosity of placebo and gels.
At low pH (about pH 3), the carbopol chains exist in a spiral coiled form, exhibiting a relatively low viscosity. With the progression of pH, the carboxyl groups of the carbopol become ionized, causing an increased repulsion of negative charges. It leads to the unwinding of molecular structure and a gradual rise in viscosity. Values of viscosities are also given in . Almost all the gels were free flowing at room temperature with little variation in viscosity with an increasing strength of P 407, and their rheological behavior was observed to be Newtonian at that temperature. As far the gelation time is concerned for the gels, it was observed to be 30 ± 2 s. Gelation time may be defined as the time when the elasticity modulus becomes higher than the viscosity modulus.
In vitro drug release kinetics
The release behavior (figure not given) could be helpful in predicting the gel’s performance in the vaginal milieu. The highest release (72.4% in 20 h) was achieved by G2, followed by G3 (66.6% in 20 h), and then G4 (62.2% in 20 h). A faster release of itraconazole expected in vivo because of the presence of salts and proteins. All the three formulations showed a sustained release profile. One unexpected phenomenon observed in the formulations was an initial burst release, which was not expected considering the presence of drug in solid lipidic core and then dispersed into gel matrix. With the progression of time, all the formulations achieved a sustained release profile. The concentration of polymer was also found to have a role in release profile. With increasing polymer (P 407) concentration (15–18%), drug release was found to be decreasing. More concentrated gels were reported to dissolve at a slower rate than less concentrated ones because of the decreased water diffusion through the gel. Dissolution of polymer is actually a controlling factor in drug release but it is not the only factor, the surface is eroding at a constant rate. It is actually a two-step process. Other than burst release, the drug molecule first oozes out from the solid lipid core then diffuse through the gel matrix. Hence considering the gelation temperature and In vitro drug release, G4 was chosen for further evaluation.
In vitro cell toxicity studies of gel (G4)
Cellular viability of HeLa-S3 cells was investigated for different concentrations of itraconazole gel in PBS. Different concentrations of G4 did not show any cytotoxicity except at a concentration of 1000 μg/ml, where cell viability decreased by about 7%. Thus, we may conclude that neither itraconazole molecule not excipients used were having any cytotoxic effect on vaginal tissues.
Preclinical evaluation of gel (G4)
Bioadhesion and irritation tests
The intensity of dye in animals containing optimized gel (G4) was much higher than control which elicits the bioadhesive potential of the formulation. The basic components of the mucus layers are mucin glycoproteins with oligosaccharide side chains. Sialic acids are located at the terminal ends of the oligosaccharide chains. Since the pKa of sialic acid is 2.6, the mucin network carries a substantial negative charge at physiological vaginal pH of pH 4–5. This charged portion interacts with the ionic components of the gel and results in appreciable bioadhesion.
As the gel is applied to the vaginal mucosa also the mechanical bioadhesion occurred with all surface voids between the gel and tissue filled and, therefore, results in surface interlocking. With the gradual hydration, a more unyielding chemically adhesive interaction progresses between the hydrated polymeric blend and the tissue interface either through ionic or covalent bonding. Weak physical forces like van der Waals and London forces are also involved in bioadhesion and once the gel diffuses through the surface, it appears a sintering-like mechanism. The strength of bioadhesion depends on a hybrid between the dispersive and diffusive phases as well as the contact surface area. The lifetime of these bonds can vary from microseconds to seconds because, at any time sufficient energy is (KT, where K is the Boltzmann constant and T is the absolute temperature) available to break these bonds. Hence, during the course of bioadhesion, these physical bonds break several times.
Optimized gel (G4) did not show any sign of toxicity. On a 0–4 scale basis, it was given the score zero, considering the negligible signs of irritation. Even after pathological examinations, it was found to be safe.
In vivo antifungal studies
A single dose of developed gel brought the microbial count to a negligible level after 21 d, whereas the marketed formulation did not bring similar amelioration in given time (Supplementary Figure 2). This also suggests that the VDDS exhibited a sustained effect in vivo. Kinetics of infection clearance was little different in initial days. Marketed formulation was able to cut down the infection at a higher pace. Reason behind may be the drug-release characteristics. SLN was not having a burst release property as the itraconazole molecules were supposed to be present in the core of particle.
Conclusion
An analysis and understanding of the available treatment stratagems for various vaginal afflictions led us to devising a model formulation based on strategies to overcome the physiological challenges offered by the target site. The purported formulation bearing SLNs loaded with itraconazole offers to assimilate itself in the vaginal milieu with prolonged muco-retention, high tolerability, and enhanced antifungal efficacy. The formulation was successfully tested in an animal infection model and is expected to deliver similar treatment outcomes in patient suffering from candidiasis.
Supplementary material available online
supplementary_3.tif
Download TIFF Image (661.5 KB)Suppl_file_2_Mirza.jpg
Download JPEG Image (79.7 KB){kind=link}
Supplementary_file_1_Mirza.jpg
Download JPEG Image (155.2 KB){kind=link}
Supplementary_TABLE.docx
Download MS Word (12.5 KB)Acknowledgements
The authors are thankful to Mr. Abrar M. Khan and Er. Asad Eqbal for their technical and logical supports and Hena Tahir for language support and preparation of manuscript. The authors are also thankful to Gattefosse (France) and BASF (Germany) for providing gift samples of different excipients.
Declaration of interest
One of the authors (M. Aamir Mirza) is grateful to University Grants Commission, New Delhi for providing financial assistance.
Related Research Data
References
- Almeida AJ, Souto E. (2007). Solid lipid nanoparticles as a drug delivery system for peptides and proteins. Adv Drug Deliv Rev 59:478–90.
- Baloglu E, Ozyazici M, Hizarcioglu SY, Karavana HA. (2003). An in vitro investigation for vaginal bioadhesive formulations: bioadhesive properties and swelling states of polymer mixtures. Farmaco 58:391–6.
- Barrett AM, Dehghani F, Foster NR. (2008). Increasing the dissolution rate of itraconazole processed by gas antisolvent techniques using polyethylene glycol as a carrier. Pharm Res 25:1274–89.
- Benziger DP, Edelson J. (1983). Absorption from the vagina. Drug Metab Rev 14:137–68.
- Bergh PA. (1988). Vaginal changes with aging, the gynaecologist and the older patient. Rockville, MD: Aspen Publishers
- Betageri GV, Makarla KR. (1995). Enhancement of dissolution of glyburide by solid dispersion and lyophilization techniques. Int J Pharm 126:155–60.
- Blanco-Fuente H, Esteban-Fernandez B, Blanco-Mendez J, Otero-Espinar FJ. (2002). Use of beta-cyclodextrins to prevent modifications of the properties of carbopol hydrogels due to carbopol-drug interactions. Chem Pharm Bull (Tokyo) 50:40–6.
- Bloch B, Barnard PG, Burger GD, et al. (1988). Itraconazole in the treatment of acute vaginal candidiasis. S Afr Med J 73:172–3.
- Choi HG, Jung JH, Ryu JM, et al. (1998). Development of in situgelling and mucoadhesive acetaminophen liquid suppository. Int J Pharm 165:33–44.
- Chopra S, Motwani SK, Iqbal Z, et al. (2007). Optimisation of polyherbal gels for vaginal drug delivery by Box-Behnken statistical design. Eur J Pharm Biopharm 67:120–31.
- De Bernardis FLVIA, Boccanera M, Adriani D, et al. (1997). Protective role of antimannan and anti-aspartyl proteinase antibodies in an experimental model of Candida albicans vaginitis in rats. Infect Immun 65:3399–405.
- Edsman K, Carlfors K, Petersson R. (1998). Rheological evaluation of poloxamer as an in situ gel for ophthalmic use. Eur J Pharm Sci 6:105–12.
- Escobar-Chavez JJ, Lopez-Cervantes M, Naik A, et al. (2006). Applications of thermo-reversible pluronic F-127 gels in pharmaceutical formulations. J Pharm Pharm Sci 9:339–58.
- Francois M, Snoeckx E, Putteman P, et al. (2003). A mucoadhesive, cyclodextrin-based vaginal cream formulation of Itraconazole. AAPS Pharm Sci 5:1–5.
- Garces R, Mancha M. (1993). One-step lipid extraction and fatty acid methyl esters preparation from fresh plant tissues. Anal Biochem 21:1139–43.
- Ge MT, Rananavare SB, Freed JH. (1990). ESR studies of stearic acid binding to bovine serum albumin. Biochim Biophys Acta 1036:228–36.
- Goppert TM, Muller RH. (2005). Polysorbate-stabilized solid lipid nanoparticles as colloidal carriers for intravenous targeting of drugs to the brain: comparison of plasma protein adsorption patterns. J Drug Target 13:179–87.
- Hancock BC, Dupuis Y, Thibert R. (1999). Determination of the viscosity of an amorphous drug using thermomechanical analysis (TMA). Pharm Res 16:672–5.
- Hwang S, Owada E, Suhardja L, et al. (1977). Systems approach to vaginal delivery of drugs IV: methodology for determination of membrane surface pH. J Pharm Sci 66:778–81.
- Jenning V, Gysler A, Schafer-Korting M, Gohla SH. (2000). Vitamin A loaded solid lipid nanoparticles for topical use: occlusive properties and drug targeting to the upper skin. Eur J Pharm Biopharm 49:211–18.
- Khanvilkar K, Donovan MD, Flanagan DR. (2001). Drug transfer through mucus. Adv Drug Deliv Rev 48:173–93.
- Levy MY, Schutze W, Fuhrer C, Benita S. (1994). Characterization of diazepam submicron emulsion interface: role of oleic acid. J Microencapsul 11:79–92.
- Mirza MA, Talegaonkar S, Ahmed FJ, Iqbal Z. (2011). A novel and multifaceted excipient for vaginal drug delivery. J Excip Food Chem 2:98–112.
- Mirza MA, Talegaonkar S, Iqbal Z. (2012). Particle engineering in the perspective of thermodynamics: a case study with Itraconazole. Adv Sci Lett 16:148–58.
- Mobley WC, Schreier H. (1994). Phase transition temperature reduction and glass formation in dehydroprotected lyophilized liposomes. J Control Release 31:73–87.
- Owen DH, Katz DF. (1999). A vaginal fluid simulant. Contraception 59:91–5.
- Paavonen J. (1983). Physiology and ecology of the vagina. Scand J Infect Dis Suppl 40:31–5.
- Repka MA, McGinity JW. (2001). Bioadhesive properties of hydroxypropylcellulose topical films produced by hot-melt extrusion. J Control Release 70:341–51.
- Ricci M, Puglia C, Bonina F, et al. (2005). Evaluation of indomethacin percutaneous absorption from nanostructured lipid carriers (NLC): in vitro and in vivo studies. J Pharm Sci 94:1149–59.
- Richardson JL, Illum L. (1992). Routes of delivery: case studies: (8) the vaginal route of peptide and protein drug delivery. Adv Drug Deliv Rev 8:341–66.
- Saag MS, Dismukes WE. (1988). Azole antifungal agents: emphasis on new triazoles. Antimicrob Agents Chemother 32:1–8.
- Schwarz C, Mehnert W. (1999). Solid lipid nanoparticles (SLN) for controlled drug delivery. II. Drug incorporation and physicochemical characterization. J Microencapsul 16:205–13.
- Song Y, Wang Y, Thakur R, et al. (2004). Mucosal drug delivery: membranes, methodologies, and applications. Crit Rev Ther Drug Carrier Syst 21:195–256.
- Tadros TF, Vincent B. (1980). Influence of temperature and electrolytes on the adsorption of poly (ethylene oxide)-poly (propylene oxide) block copolymer on polystyrene latex and on the stability of the polymer-coated particles. J Phys Chem 84:1575–80.
- Trott O, Olson AJ. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–61.
- Wang Y, Lee CH. (2002). Characterization of a female controlled drug delivery system for microbicides. Contraception 66:281–7.
- Washington C. (1996). Stability of lipid emulsions for drug delivery. Adv Drug Deliv Rev 20:131–45.
- Weuts I, Kempen D, Six K, et al. (2003). Evaluation of different calorimetric methods to determine the glass transition temperature and molecular mobility below T(g) for amorphous drugs. Int J Pharm 259:17–25.
- Yoo SD, Kang E, Jun H, et al. (2000). Absorption, first-pass metabolism, and disposition of itraconazole in rats. Chem Pharm Bull (Tokyo) 48:798–801.
- Zaki NM, Awad GA, Mortada ND, Abd Elhady NM. (2007). Enhanced bioavailability of metoclopramide HCl by intranasal administration of a mucoadhesive in situ gel with modulated rheological and mucociliary transport properties. Eur J Pharm Sci 32:296–307.