
Abstract
Context: Pravastatin sodium (PVS) is a freely water-soluble HMG-CoA inhibitor that suffers from instability at gastric pH, extensive first pass metabolism, short elimination half-life (1–3 h) and low oral bioavailability (18%). Objective: To overpower these drawbacks and to maximize drug absorption at its main site of absorption at the duodenum, enteric surface-coated PVS-loaded nanocubosomal dispersions were presented. Materials and methods: Glyceryl monooleate (GMO)-based dispersions were developed by the fragmentation or the liquid precursor methods using Pluronic® F127 or Cremophor® EL as surfactants. As a challenging enteric-coating approach, the promising dispersions were surface-coated via lyophilization with Eudragit® L100-55; a duodenum-targeting polymer. The drug content, particle size, zeta potential, morphology and release studies of PVS-loaded dispersions were evaluated before and after surface-coating. Compared to an aqueous PVS solution, the pharmacokinetics of the best achieved system (E-F8) was evaluated (UPLC-MS/MS) in rats. Results: The enteric surface-coated nanocubosomal dispersions were more or less spherical in shape and showed high drug-loading, negative zeta potential values and fine-tuned biphasic drug-release patterns characterized by retarded (2 h) and sustained (10 h) phases in pH 1.2 and pH 6.8, respectively. E-F8 system showed significantly (p< 0.05) higher oral bioavailability, delayed Tmax and prolonged MRT0−∞ following oral administration in rats. Conclusions: The duodenum-triggering potential and the controlled-release characteristics of the best achieved system for smart PVS delivery were revealed.
Introduction
The use of free drugs in conventional dosage forms generally involves difficulties in achieving the target site at the appropriate doses after or during a proper time period. Consequently, smart drug delivery to specific organs and tissues has become one of the critical endeavors to address their physicochemical and pharmacokinetic characteristics, while improving patient compliance (Sastry et al., Citation2000; Martinho et al., Citation2011).
Drug-loaded lipid-based systems like liposomes, solid lipid nanoparticles, nanostructured lipid carriers and cubosomes have become an outstanding theme of research in therapeutics (Salunkhe et al., Citation2015; Talluri et al., Citation2015). The self-assembly of amphiphilic lipids into well-defined thermodynamically stable structures [micellar (LI), hexagonal (HI), cubic (QII), lamellar (Lα), inverted micellar (LII) and inverted hexagonal (HII)] upon exposure to a polar medium was previously reported and investigated for effective drug targeting (Ganem-Quintanar et al., Citation2000). Amongst these structures, the self-assembled cubic liquid crystalline nanoparticles (cubosomes) were emerged as one of the most versatile, scalable, cost-effective and clinically promising technology platforms. They possess a unique nanostructure consisting of highly twisted continuous lipid bilayers and two congruent non-intersecting water channels. This complex structure has been postulated to generate lipophilic and hydrophilic domains to host bioactives (Rizwan et al., Citation2011). The hydrophilic compounds can be enveloped within the water channels, the lipophilic ones can be loaded into the lipid bilayers while the amphiphilic molecules may partition at the lipid/water-interface. Compared with liposomes, the high bilayers surface area to volume ratio of cubosomes can increase the relative payload of drugs (Siekmann et al., Citation2002). Furthermore, cubosomes can potentially control the rate of drug release and protect it against physiological and/or chemical degradation (Ganem-Quintanar et al., Citation2000; Kwon et al., Citation2012).
The unsaturated long chain monoglycerides, like Glyceryl monooleate (GMO), are nontoxic, biocompatible and biodegradable amphiphilic molecules. In aqueous milieu, GMO can spontaneously form reverse bicontinuous cubic phases (optical transparent gels) due to the isotropic structure of GMO (Mariani et al., Citation1988). Practically, the administration of a drug that is suspended within a GMO matrix is inconvenient due to the latter's semisolid and sticky nature. Two approaches were investigated to surmount this restriction; the first one involves the fragmentation of the cubic phase into cubosomes using aqueous surfactant, like Pluronic® F127, solutions. The alternative technique depends on the use of liquid precursors where polar co-solvents, like ethanol or PEG 400, are added to GMO-surfactant phases allowing for the spontaneous fabrication of cubosomes upon contact with excess water (Moebus et al., Citation2012).
Pravastatin sodium (PVS) is a cholesterol-lowering statin that acts on 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase enzyme through a reversible inhibition mechanism in animals and humans (Komai et al., Citation1992). Following oral administration, PVS is mainly absorbed from the duodenum and rapidly reaches its maximum drug-plasma concentration within approximately one hour. From a clinical point of view, PVS suffers from low oral bioavailability (18%) and short elimination half-life of PVS (1 to 3 h). This obligates frequent dosing to maintain a prolonged drug-plasma concentration that is suitable for daily dosing (Hatanaka, Citation2000). Previous reports on PVS related its low oral bioavailability to many factors. The first one is related to the marked instability in the stomach and possible transformation, via non-enzymatic acid-catalysed isomerization, to 3′α-isopravastatin and 6′-epipravastatin. Compared to other statins, the intrinsic hydrophilic properties of PVS (octanol/water partition coefficient of 0.59 at pH 7.0) limit its permeability and absorption through the intestinal membranes. Furthermore, PVS undergoes extensive first pass metabolism; up to 50% of dose is substantially metabolized by liver before reaching the systemic circulation (Singhvi et al., Citation1990; Quion & Jones, Citation1994).
From the authors’ point of view, the development of duodenum-triggered nano-cubosomal systems could recoil PVS downsides. In fact, the design of promising enteric surface-coated carriers has gained considerable attention in the last years (Chandy et al., Citation2002; Eerikäinen & Kauppinen, Citation2003; Bejugam et al. Citation2008; Zhang et al., Citation2011; Hao et al., Citation2013). Yet, most of the reported methods suffer from many limitations including the lengthy production steps, the use of organic solvents, the need of special sophisticated equipment and/or the limited scaling up possibilities. In a previous work (Tayel et al., Citation2015), the authors developed a challenging surface-coating, duodenum-triggered, approach for PVS-loaded spanlastics using Eudragit® L100-55. Based on the adopted freeze drying technique, no organic solvent was needed and the possibility of scaling-up could be achieved in a single-step. Herein, the feasibility of this approach for cubosomes, as another PVS carrier system, was explored. Several nanocubosomal systems were fabricated by the fragmentation and the liquid precursor methods to select an appropriate method of preparation. The systems were evaluated with respect to drug content, particle size, zeta potential, morphology and PVS release profiles before and after surface-coating. Finally, the duodenum-triggering potential and the controlled-release characteristics of the best achieved system were explored in rats by UPLC-MS/MS.
Materials and methods
Materials
PVS and rosuvastatin calcium (RVC) were donated by Hi-Pharm (Obour city, Egypt) and Chemipharm, (6th October city, Egypt), respectively. Glyceryl monooleate (GMO) was a grant from Abitec Corporation (Columbus, OH). Eudragit® L100-55 was provided by Rohm GmbH & Co. KG (Darmstadt, Germany). Spectra Por© semi-permeable membrane tubing (MWCO 12,000–14,000) was obtained from Spectrum Laboratories Inc., (Rancho Dominguez, CA). Pluronic® F127 and Cremophor® EL were purchased from BASF Chemical Company (Ludwigshafen, Germany). Trehalose, methanol (HPLC grade), formic acid solution (HPLC grade) and o-phosphoric acid solution (HPLC grade) were purchased from Sigma Chemical Co. (St. Louis, MO). Potassium dihydrogen phosphate and disodium hydrogen phosphate were purchased from Merck (Darmstadt, Germany). Polyethylene glycol (PEG) 400, chloroform, ethanol (95%) and concentrated hydrochloric acid were acquired from El-Nasr pharmaceutical chemicals Co. (Cairo, Egypt). All other chemicals were of analytical grade and were used as received.
Preparation of GMO-based cubosomal dispersions via the fragmentation method
Aqueous solutions of PVS were warmed (40 °C) and added in a drop-wise manner to molten (40 °C) GMO. The mixtures were kept under magnetic stirring (300 rpm) for 30 min. The immediately formed viscous gels were left to equilibrate in screw capped, tightly sealed, glass vials at room temperature (25 ± 0.5 °C) for 24 h. The formation of the isotropic cubic phases was optically verified through the formation of transparent gels (Esposito et al., Citation2003; Wörle et al., Citation2007).
Following, the gels were fragmented (dispersed) by the addition of aqueous surfactant solutions (2%, w/w) while being mechanically stirred. To promote the development of fine opalescent cubosomal dispersions, the crude systems were homogenized using an Ultra-Turrax homogenizer (IKA Labortechnik, Staufen, Germany) for 5 min at 30,000 min−1. The investigated variables include; (i) the GMO: PVS ratio; 10:1 and 20:1, (ii) the surfactant type; Pluronic® F127 and Cremophor® EL, and (iii) the GMO: surfactant ratio; 2.5: 1 and 5: 1. The composition of the investigated dispersions is shown in .
Table 1. The composition (g) of the investigated PVS-loaded cubosomal dispersions prepared by the fragmentation method.
Preparation of GMO-based cubosomal dispersions via the liquid precursor method
Accurately weighed amounts of GMO, surfactant and hydrotrope (PEG 400) were dissolved in chloroform and left on a magnetic stirrer (300 rpm) overnight to allow complete evaporation of the organic phase. The resulting liquid precursors were dispersed into aqueous solutions of PVS while being mechanically stirred for 30 min. To promote the development of fine opalescent cubosomal dispersions, the crude systems were vortexed for 10 min (Kojarunchitt et al., Citation2011). The investigated variables include; (i) the GMO: PVS ratio; 10: 1 and 20: 1 (ii) the surfactant type; Pluronic® F127 and Cremophor® EL, and (iii) the hydrotrope: (GMO – surfactant) ratio; 1.5: 1 and 3: 1. The composition of the investigated dispersion is shown in .
Table 2. The composition (g) of the investigated PVS-loaded cubosomal dispersions prepared by the liquid precursor method.
In vitro characterization of PVS-loaded cubosomal dispersions
Investigation of the physicochemical interactions between PVS and GMO
The PVS-loaded cubic phase gels were frozen and lyophilized (24 h, −45 °C) using Novalyphe-NL 500 lyophilizer (Savant Instruments; NY, USA) under a pressure of 7 × 10−2 mbar. Pure PVS powder and the lyophilized gels were subjected to the following solid-state investigations.
Fourier transform infrared spectroscopy
The Fourier transform infrared spectroscopy (FT-IR) spectra of the samples were scanned using Affinity-1 spectrophotometer (Shimadzu, Kyoto, Japan) over the spectral region of 4000 to 400 cm − 1 to explore the possible chemical intermolecular interactions between PVS and GMO. The detector was purged with clean dry nitrogen gas in order to increase the signal sensitivity. The samples were prepared according to the potassium bromide disc technique. Briefly, the samples were mixed with potassium bromide (1:10, w/w) and pressed to form a disc. Since the potassium bromide has no absorption in the fundamental FT-IR spectrum region, only the spectrum of the investigated sample is obtained (Tayel et al., Citation2013).
Differential scanning calorimetry
The differential scanning calorimetry (DSC) thermograms of the samples were recorded on a differential scanning calorimeter (DSC-60, Shimadzu, Kyoto, Japan) to describe their thermotropic properties and to estimate their degree of crystallinity and/or the possible incompatibilities between PVS and GMO. Briefly, three to four milligram samples were heated in hermetically sealed flat-bottom aluminum pans over a temperature range of 30–200 °C, at a rate of 10 °C/min, under a nitrogen purge of 30 ml/min (Ghorab et al., Citation2011).
Drug content of dispersions
With the aim to quantify PVS content of dispersions after production, a sample of dispersion was appropriately diluted in ethanol and then sonicated for 10 min till a clear solution is obtained. The drug content was determined spectrophotometrically (Shimadzu UV-1601 PC; Kyoto, Japan) at λmax 239 nm. The drug content percentages were calculated according to EquationEquation (1)(1) (Morsi et al., Citation2014);
(1)
The calculated amount of PVS was taken as reference of the total amount of drug for further studies (Esposito et al., Citation2005). The results were statistically analyzed using Design-Expert® software (Stat-Ease, Inc., Minneapolis, MN, USA) as a (22.22) full factorial design. Comparisons were evaluated using one-way ANOVA at a p value < 0.05.
Determination of particle size, zeta potential and polydispersity index
The mean hydrodynamic diameter (z-average) and the polydispersity index (PI) of PVS-loaded nanocubosomal dispersions were determined using a Zetasizer Nano ZS (Malvern instruments; Worcestershire, UK) according to the Dynamic Light Scattering (DLS) technology. A wavelength of 635 nm was selected while the scattering angle was fixed at 90°. Based on the Electrophoretic Light Scattering (ELS) technology, the zeta potential (ζ) values of the dispersions were estimated using a Laser Doppler Anemometer coupled with the same equipment.
Measurements were carried out at room temperature (25 ± 0.5 °C) after appropriate dilution of the samples with de-ionized water to adjust the signal level. The average of three successive measurements of three independent samples was calculated.
In vitro drug release studies
Taking into consideration the expected increase in size following the enteric surface-coating process, a constraint of vesicle size was needed. Consequently, the cubosomal dispersions having z-average values > 150 nm were excluded from further studies (Tayel et al., Citation2015).
The in vitro release studies of PVS from an aqueous solution (20 mg/ml; control formula) and from the fine cubosomal dispersions (< 150 nm), were carried out in triplicate, within a USP Dissolution Testing Station, Type II (VK 7000, Vankel Industries, Inc., NJ, USA) at 37 ± 0.5 °C. Based on the drug content percentages, a sample of each dispersion containing the equivalent to 20 mg of PVS was loaded, well sealed in Spectra Por© semi-permeable membrane tubing and then immersed into the dissolution vessel. The dissolution medium was de-aerated water (900 ml) and the rotation speed of the paddles was set at 50 rpm (Niazi, Citation2014). Aliquot samples (4 ml) were withdrawn at certain time intervals up to 8 h and were immediately replaced with an equal volume of fresh medium to maintain a fixed volume. The drug released percentages were determined as previously described at λmax 239 nm. Statistical analysis via SPSS software (SPSS Inc., Chicago, USA) was evaluated for the drug released percentages after 2 h (P2h) and 8 h (P8h) using one way ANOVA test at p < 0.05.
Preparation of enteric surface-coated cubosomal dispersions
Eudragit® L100-55 is an enteric polymer that dissolves above pH 5.5 and releases its cargo at the duodenum. The aptitude of Eudragit® L100-55 as a surface-coating polymer was explored for the selected fine cubosomal dispersions.
According to the method developed by Tayel et al. (Citation2015), a sample of each dispersion containing the equivalent to 200 mg of PVS was sonicated (5 min) in 100 ml Sorensen’s phosphate buffer (pH 6.8) to which Eudragit® L100-55 was previously dissolved at three concentrations; 0.25, 0.50 or 0.75%, w/v. Following, trehalose (10%, w/v) was incorporated as a cryoprotectant. The resulting systems were frozen, lyophilized and reconstituted in Sorensen’s phosphate buffer (pH 5) to avoid the possible dissolution of the enteric surface-coat at higher pH values (Leroux et al., Citation1995).
In vitro characterization of enteric surface-coated cubosomal dispersions
The enteric surface-coated cubosomal dispersions were characterized for particle size, polydispersity index, zeta potential as previously mentioned. The in vitro drug release studies were conducted on dispersion samples containing the equivalent to 20 mg of PVS. The studies were conducted in 0.1N HCl (pH 1.2, 2 h) and then in Sorensen’s phosphate buffer (pH 6.8, 10 h). Statistical analysis of the drug released percentages after 2 h (P2h) and 12 h (P12h) was tested using one way ANOVA test at p < 0.05. TEM micrographs were captured to investigate the topography of representative dispersions.
In vivo absorption studies of PVS in rats
Study design
The in vivo absorption studies of the best achieved PVS-loaded nanocubosomal dispersion (E-F8), relative to an aqueous drug solution, were carried out in male Wister rats (200 ± 20 g) to provide a preliminary indication of the intrinsic impact of the cubic phase nanoparticles on drug absorption. The animal experiments followed a two-treatment, non-blind, randomized, parallel design. The studies were reviewed, approved (PI 1195) and conducted in accordance with the guidelines of the institutional Research Ethics Committee (REC-FOPCU) as well as the EU Directive 2010/63/EU for animal experiments.
The rats were cared for and handled according to the institutional regulations for experimental animals. They were housed in an animal care facility under standardized conditions of controlled temperature (25 ± 0.5 °C), humidity (50 - 60%) and alternate 12 h light–dark cycles. They were kept on standard pellet diet while water was made available ad libitum. These measures were taken to minimize the factors that can impact the animal welfare and/or influence the reproducibility and validity of the derived preclinical data.
Oral administration of treatments and sample collection
Ten adult male Wistar rats were randomly assigned to one of two groups of equal number. On the study day, the overnight (12 h) fasted rats of group I were orally treated with an aqueous PVS solution (treatment A) equivalent to 20 mg/kg while those of Group II were orally administered with E-F8 cubosomes (treatment B) at the same dose (Kivistö et al., Citation2005).
Blood samples were collected, under mild ether anesthesia, prior to and at intervals up to 8 h after oral administration of treatments. The samples were immediately placed into 1.5 ml micro-centrifuge tubes containing heparin sodium and centrifuged for 5 min at 5000 g. One hundred and fifty microliters of the plasma samples were separated, kept in labeled glass tubes and frozen at −20 °C till analysis.
Sample processing
The plasma samples were acidified (15 μL of ortho-phosphoric acid solution; 10%, v/v) and spiked with the internal standard solution (10 μL of RVC solution, 100 ng/ml). The treated samples were vortex mixed and then extracted with a mixture of diethyl ether and dichloromethane (1 ml, 70: 30). The samples were vortex-mixed and centrifuged to separate the organic layers. The latter were transferred into Wassermann tubes, positioned in a vacuum concentrator (Eppendorf 5301; Hamburg, Germany) and evaporated to dryness at 45 °C. The dry residues were reconstituted and vortex mixed with 50 μl of the mobile phase. Five microliters of the samples were injected via the auto-sampler for analysis.
Determination of PVS in rat plasma by UPLC–MS/MS
The ultra-performance liquid chromatography (UPLC) was performed on an ACQUITY UPLC™ system (Waters Corp., Milford, MA) fitted with a binary solvent delivery system and a sample manger coupled to ACQUITY™ TQD 6420 triple quadrupole tandem mass spectrometer. The latter is equipped with TurboionsprayTM interface and electrospray ionization (ESI) source.
The determination of PVS in rat plasma was based on a slightly modified sensitive, selective and previously adopted UPLC-MS/MS method (Ma et al., Citation2014; Tayel et al., Citation2015). Briefly, the chromatographic separation was performed, at 25 °C, on an ACQUITY UPLC™ BEH C18 column (1.7 μm; 150 × 2.1 mm) with a mobile phase consisting of methanol: 0.1% formic acid [80: 20] at a flow rate of 0.3 ml/min. The ESI source was adjust to operate in the positive ion mode. The detector was tuned to operate in the multiple reaction monitoring (MRM) mode to detect the transitions of the m/z 482.20 precursor ion to the m/z 258.17 product ion for RVC and the m/z 447.35 precursor ion to the m/z 327.23 product ion for PVS. The UPLC-MS/MS data acquisition and integration were processed using MassLynx™ Software Ver. 4.1 SCN 805 (Waters Corporation; MA, USA). The total run time was 2.0 min only; allowing for the simultaneous detection of RVC and PVS at 1.26 and 1.28 min, respectively.
Pharmacokinetic and statistical analyses
The drug-plasma concentration time profiles for each rat and the mean drug concentration for each dose group within each treatment were analyzed with WinNonlin® software (Scientific consulting Inc., NC, USA) following a non-compartmental analysis module. The relevant pharmacokinetic parameters were estimated, including; the peak PVS plasma concentration (Cmax, ng/ml), the time elapsed to reach PVS peak concentrations (Tmax, h), the mean residence time from zero to infinity (MRT(0−∞), h) and the area under the drug-plasma concentration time curve from zero to infinity AUC(0−∞), ng.h/mL). The increase in the oral bioavailability (folds) was assessed from the AUC(0−∞) of both treatments. The data were statistically analyzed using ANOVA with Post Hoc multiple comparisons to assume the statistical significance at p < 0.05.
Results and discussion
Preparation of GMO-based cubosomal dispersions
Two approaches were investigated to develop GMO-based cubic liquid crystalline nanoparticles; cubosomes. The first approach depends on the fragmentation of the stabilized bulk cubic gels by the addition of an aqueous surfactant solution. Meanwhile, the second technique allows for the spontaneous formation of nanoparticles upon dilution of the liquid precursor system.
GMO is a nontoxic, biodegradable, and biocompatible polar lipid that possesses a structure resembling nonionic surfactants. The glycerol moiety (the polar head) has active hydroxyl groups and can form hydrogen bonds with aqueous milieu. On contrary, the oleic acid hydrocarbon chains (the tail) impart the hydrophobic characteristics (Ganem-Quintanar et al., Citation2000). The previously evaluated phase behavior for GMO – water systems revealed that at 40 °C and in the presence of a small amount of water, GMO forms reversed micelles (LII). Adding more water would promote the formation of the lamellar phase (Lα) while a large isotropic and highly viscous cubic phase predominates when the water percentage is raised to 20–40% (Clogston et al., Citation2000; Ganem-Quintanar etal., 2000).
For the development of cubosomes via the fragmentation method, in the current work, a fixed concentration of GMO (65%) and an aqueous drug solution (35%) was selected as an appropriate composition to ensure complete solubilization of PVS in water (solubility of PVS in water is higher than 300 mg/ml) and be close to the excess water boundary in the phase diagram. This would allow the development of the desired cubic phase (Pn3m) and minimize further water uptake on exposure to the receptor medium (Lee et al., Citation2009)
In vitro characterization of PVS-loaded cubosomal dispersions
Investigation of the physicochemical interactions between PVS and GMO
FT-IR studies
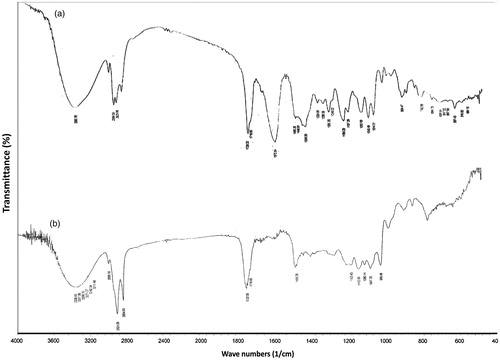
The FT-IR spectra of PVS and a representative PVS-loaded lyophilized cubic phase gel (F8) were scanned to explore the possible intermolecular interactions between PVS and GMO. As depicted in , the FT-IR spectrum of PVS showed a broad band at 3390 cm − 1 that could be attributed to the O–H stretching vibrations. The bands at 2954 and 2935 cm − 1 could be assigned to C–H stretching vibrations of methyl and/or methylene groups. The characteristic sharp band at 1728 cm − 1 is correlated with C = O stretching of the carboxylate group. The permanence of these characteristic bands in the FT-IR spectrum of PVS-loaded lyophilized cubic gel, , highlights the lack of considerable intermolecular interactions between PVS and GMO.
Figure 1. FT-IR spectra of PVS (a) and the lyophilized cubosomal dispersion F8 (b).
DSC studies
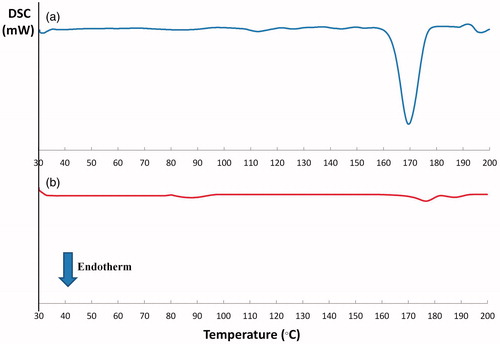
DSC thermograms of PVS and the same PVS-loaded lyophilized cubic phase gel (F8) were recorded to screen the thermotropic properties and explore the possible interactions between PVS and GMO. The DSC thermogram of PVS, , describes a typical crystalline substance characterized by one sharp endothermic peak corresponding to the melting point of the drug at 170 °C (Tayel et al., Citation2015). On contrary, the complete disappearance of this peak at the aforementioned temperature and the formation of a small broad peak at 176.30 °C in the DSC thermogram of PVS-loaded lyophilized cubic gel, , could point out the possible dispersion of PVS in an amorphous state throughout the cubic phase gel.
Figure 2. DSC thermograms of PVS (a) and the lyophilized cubosomal dispersion F8 (b).
Drug content percentages
The complex structure of cubosomes has been postulated to generate lipophilic and hydrophilic domains to host bioactives (Rizwan et al., Citation2011). The hydrophilic molecules, like PVS, can be enveloped in the two congruent non-intersecting water channels within cubosomes. In the current work, the drug content of dispersions was determined in order to make sure that the loaded amount of PVS is present in the cubosomal dispersions. It was clear that the developed dispersions, , had high drug content percentages ranging from 91.34% (F1) to 96.56% (L7); with respect to the loaded drug amount. These findings were in line with those reported for silver sulfadiazine-loaded cubosomes (Morsi et al., Citation2014) and for indomethacin-loaded cubosomes (Esposito et al., Citation2005). These percentages should be taken as reference of the total drug amount for further studies.
Table 3. The physicochemical properties of the investigated PVS-loaded nanocubosomal dispersions (mean ± S.D., n = 3).
Particle size, zeta potential and polydispersity index measurements
The mean particle size, zeta potential and PI values of the developed cubosomal dispersions are summarized in . It was clear that both methods succeeded, under the investigated conditions, to develop nanocubosomal dispersions with an average size ranging from 112.10 (F6) nm to 403 nm (L8). The ANOVA results confirmed the dependance of the particle size on the GMO: PVS ratio, the surfactant type, and the GMO:surfactant ratio (for the fragmentation method-derived dispersions) or the hydrotrope: (GMO – surfactant) ratio (for the liquid precursor method-derived dispersions).
Regardless of the method of preparation, a direct correlation was observed between the GMO:PVS ratio and the particle size. The cubosomal dispersions prepared at a GMO: PVS ratio of 20: 1 were significantly (p < 0.05) larger in size than the corresponding dispersions prepared at a GMO: PVS ratio of 10: 1. The development of highly twisted continuous thicker lipid bilayers within the former dispersions could be a possible explanation to this behavior. Larger PI values were observed with these dispersions; indicating wider particle size distributions. These findings were matched with those reported by Esposito et al. (Citation2003) showing that the mean diameter and PI values of the dispersions were increased from 193.5 nm and 0.18 to 241 nm and 0.25 on increasing the GMO-based dispersed phase concentrations from 5% to 10% (w/w), respectively.
The incorporation of a surfactant (Pluronic® F127 or Cremophor® EL) was necessary to ensure stabilization of the systems. Preliminary studies had showed that the GMO: surfactant ratio of 5: 1 was necessary to form milky crude dispersions with no apparent aggregates or phase separation post preparation. Pluronic® F127 is a nonionic surfactant composed of polyethylene oxide (PEO)-polypropylene oxide (PPO)-polyethylene oxide (PEO) block copolymer. The stabilizing effect of Pluronic® F127 is attributed to the balance between the hydrophobic domain size of PPO that is responsible for the anchoring to lipid bilayers and the hydrophilic chain length of PEO that is oriented towards the aqueous medium and acts as a steric barrier to prevent aggregation of nanoparticles (Chong et al., Citation2012). In a parallel line, Cremophor® EL is a nonionic surfactant composed of a hydrophobic tail (glycerol polyethylene glycol ricinoleate and other fatty acid esters of polyethylene glycol) and a hydrophilic head (free polyethylene glycols and ethoxylated glycerol). Similar to Pluronic® F127, the stabilizing effect of Cremophor® EL is related to the balance between the hydrophilic and the hydrophobic moieties (Madheswaran et al., Citation2014).
Pluronic® F127-based dispersions showed smaller mean particle diameters than the corresponding Cremophor® EL-based dispersions developed by the liquid precursor method. The higher HLB value (22) of Pluronic® F127, compared to that of Cremophor® EL (12-14), could prove the higher emulsification power and consequently, the stronger ability to form finer particles. On contrary, the addition of the aqueous surfactant solution following the development of PVS-loaded cubosomes in the fragmentation method might reduce the surfactant ability to form fine particles. This effect was more pronounced with Pluronic® F127-based dispersions.
In this context, two GMO: surfactant ratios (2.5: 1 and 5: 1) were investigated during the development of cubosomes by the fragmentation method. Regardless of the surfactant type, an inverse relationship was established between the surfactant concentration and the mean particle diameter. At a GMO: surfactant ratio of 5:1, the surfactant molecules might be unable to cover the entire particle surface allowing for the aggregation of some particles till their surface area is decreased to a point that the limited surfactant concentration could coat the entire agglomerate surface and consequently, form a stable dispersion with relatively larger particles (Ghorab et al., Citation2011). The higher surfactant concentration, with respect to a fixed concentration of GMO, would allow for the partitioning of more surfactant molecules into the interfacial domain of the liquid crystalline phase. This results in more steric stabilization and lower tendency for aggregation (Madheswaran et al., Citation2014).
A hydrotrope is a molecule with hydrophobic and hydrophilic character but incapable of displaying a typical surfactant behavior. Perversely, hydrotropes like PEG 400 exhibit a “salting-in” behavior by increasing the solubility of lipids such as GMO and create stable liquid precursors that can spontaneously form cubosomes when diluted (Spicer & Hayden, Citation2001). Two hydrotrope: (GMO – surfactant) ratios were investigated in the liquid precursor-derived cubosomes. There was a significant (p < 0.05) trend towards a decrease in the particle size and PDI with increasing PEG 400 concentration (Rizwan et al., Citation2011). The higher hydrotrope concentrations would lower the viscosity of the liquid precursor and may therefore aid in the subsequent dispersion by reducing the energy input necessary to produce finer particles (Spicer & Hayden, Citation2001).
The freshly prepared dispersions bear negative Zeta potential values ranging from −14.13 mV (F2) to −38.30 mV (F8). These values relatively indicate the strong electric repulsion between particles and the lowered tendency towards aggregation. A part of these negative charges could be correlated to PVS that possesses an alkyl chain terminated with a carboxylic acid group and bearing two hydroxyl groups at the β and δ positions (Nigović & Vegar, Citation2008). In addition to this constant contribution, the ANOVA results revealed the presence of direct correlations between the zeta potential values and the concentration of GMO and/or surfactants. This could be attributed to the ionization of the negatively charged carboxylic groups of the fatty acid moieties within GMO, Pluronic® F127 or Cremophor® EL.
In vitro drug release studies
The in vitro release profiles of PVS-loaded fine cubosomal dispersions prepared by the fragmentation method ((F2, F4, F6 and F8; ) and by the liquid precursor method (L1, L2, L5 and L6; ) were compared to that of an aqueous PVS solution. It was clear that almost 100% of PVS was released from the solution within 3 h; pointing out that the dialysis membrane did not held back PVS diffusion.
Figure 3. In vitro release profiles of PVS-loaded fine cubosomal dispersions prepared by the fragmentation method and an aqueous drug solution in water at 37 ± 0.5 °C (mean ± S.D., n = 3).
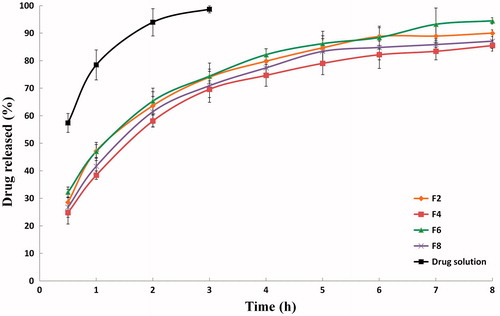
Figure 4. In vitro release profiles of PVS-loaded fine cubosomal dispersions prepared by the liquid precursor method and an aqueous drug solution in water at 37 ± 0.5 °C (mean ± S.D., n = 3).
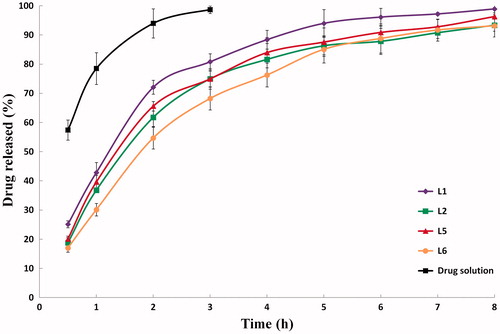
The drug released percentages after 2 h (P2h) and 8 h (P8h), shown in , were statistically checked to highlight the significant differences between the investigated drug release profiles. To attain promising controlled-drug release patterns, lower P2h and higher P8h percentages are desired. The initial release phase (P2h) might be attributed to the release of the un-entrapped and/or the surface-adsorbed drug while the sustained release phase (P8h) reflects the release of PVS from the water channels within cubosomes. It seems that the drug snakes his way through the water channel network and eventually reaches the sink dissolution medium.
The statistical analysis of the drug release data of fine cubosomal dispersions confirmed that P2h and P8h are dependent on some of the factors influencing the particle size, including; (i) the GMO: PVS ratio, (ii) the surfactant type and (iii) the hydrotrope: (GMO – surfactant) ratio. These findings stem from the fact that larger cubosomes would increase the drug diffusional distance and consequently slow down the drug dissolution rates (Wacker, Citation2013). It was clear that significantly (p < 0.05) higher drug released percentages (P2h and P8h) were achieved when the cubosomes were prepared at the lower GMO: PVS ratio (10: 1) and the higher hydrotrope: (GMO – surfactant) ratio (3: 1). As noted before, Pluronic® F127-containing dispersions showed higher P2h and P8h with the liquid precursor method-derived cubosomes and lower P2h and P8h with the fragmentation method-derived cubosomes.
Preparation and preliminary evaluation of enteric surface-coated cubosomal dispersions
The fine cubosomal dispersions were surface-coated with Eudragit® L100-55 to endorse the delivery of PVS to the duodenum. Eudragit® L 100-55 is an anionic pH-dependent copolymer of methacrylic acid and ethyl acrylate. This structure allows for the dissolution of the polymer above pH 5.5 where the duodenum would be the initial drug release region. In view of the aforementioned, a new application for the lyophilization technique was explored to develop enteric surface-coated nanocubosomal dispersions in a single-step and without the need of organic solvents.
Preliminary studies were conducted on one representative nanocubosomal dispersion (F8) using three concentrations of Eudragit® L100-55 solutions viz. 0.25, 0.50 and 0.75% (w/v). The developed enteric-coated nanocubosomal dispersions were evaluated for particle size and in vitro drug release studies to select the optimum polymer concentration.
A direct correlation was observed between the size of the enteric surface-coated cubosomes and the concentration of Eudragit® L100-55 solution. The mean size of F8 cubosomes (118.60 nm) was significantly (p < 0.001) increased to 432.60, 523.60 and 583.30 nm, respectively. It could be deduced that higher Eudragit® L100-55 concentrations (0.50 and 0.75%, w/v) would produce more viscous solutions capable of generating thicker coats surrounding the particles (Maghsoodi, Citation2009).
The in vitro release profiles of PVS-loaded cubosomal dispersion (F8) coated with different concentrations of Eudragit® L100-55 in 0.1N HCl (pH 1.2, 2 h) and in Sorensen’s phosphate buffer (pH 6.8, 10 h) at 37 ± 0.5 °C are depicted in . According to the United States Pharmacopeia (Citation2013) specifications for enteric-coated formulations, the P2h should not exceed 10%. Herein, the observed P2h percentages were 12.32, 1.18 and 0.88%, respectively. It is obvious that the lowest concentration of the polymer solution (0.25%, w/v) failed to retard the drug released percentages in the acidic milieu; possibly due to its lower ability to provide a sufficient coating barrier. On contrary, the P12h percentages were 82.51, 67.34 and 48.49%, respectively. These significant (p < 0.01) variations stem from the use of different polymer concentrations. The highest concentration of the polymer solution (0.75%, w/v) allowed for the formation of very thick coats that lead to significant (p < 0.001) reductions in the drug released percentages over 12 h. Similar observations were noted by Hao et al. (Citation2013), for Eudragit® L100-55 nanoparticles loaded with omeprazole. It was concluded that an inverse correlation could be established between the polymer concentration and the drug release rate. At lower polymer concentrations, higher water percentages can escape prior to the solidification and the formation of coherent film-like membrane surrounding the particles. Increasing the number of pores will allow for easier water penetration and consequently higher drug released percentages. On contrary, shorter times would be required at higher polymer loads to form the surrounding coherent coats. As a consequence, lower water percentages might be allowed to diffuse into the coated particles allowing for the release of lower drug percentages (Maghsoodi, Citation2009).
Figure 5. In vitro release profiles of PVS-loaded cubosomal dispersion (F8) coated with different concentrations of Eudragit® L100-55 in 0.1N HCl (pH 1.2, 2 h) and in Sorensen’s phosphate buffer (pH 6.8, 10 h) at 37 ± 0.5 °C (mean ± S.D., n = 3).
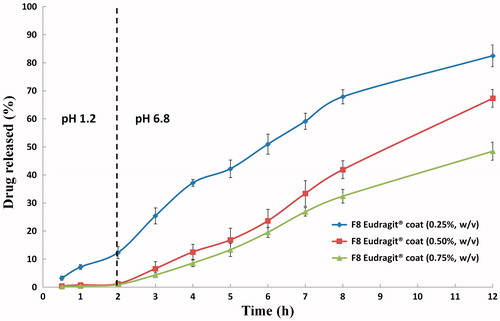
It could be inferred that a good balance between P2h and P12h could be achieved with a coating polymer solution of 0.50% (w/v). These findings and suggestions were in line with those reported in a previous work for a similarly-developed enteric surface-coated PVS-loaded spanlastics. An optimum concentration of Eudragit® L100-55 (0.5% w/v) was adopted for coating the remaining fine cubosomal dispersions.
In vitro characterization of enteric surface-coated cubosomal dispersions
The enteric surface-coated cubosomal dispersions were characterized for particle size, polydispersity index, zeta potential, morphology and in vitro drug release studies.
The increase in z-average diameters, PI values as well as ζ potential values are listed in . The mean diameters of the coated cubosomes were significantly increased (p < 0.001), compared with those of the corresponding uncoated cubosomes described in . The increase in the mean particle size percentages ranged from 280.16% (E-F2 vs F2) to 350.88% (E-F6 vs F6). For the investigated dispersions, the mean (± S.D.) increase in particle size percentage was 316.97 ± 25.84% while the CV% was 8.15%.
Table 4. The physicochemical properties of the investigated PVS-loaded enteric surface-coated cubosomal dispersions (mean ± S.D., n = 3).
The mean PI values of the coated cubosomes varied significantly (p < 0.001) from those of the corresponding uncoated cubosomes with a mean (± S.D.) increase in PI of 63.99 ± 22.42%. According to Bejugam et al. (Citation2008), the particle size distributions of the enteric surface-coated particles are potentially influenced by the presence of other types of particles along with the targeted surface-coated particles. In other words, the enteric surface-coated dispersions may include small percentages of only drug nanoparticles, only Eudragit® L100-55 nanoparticles as well as drug-Eudragit® L100-55 nanoparticles.
The significant (p < 0.01) change in the Zeta potential values following the surface-coating process may be related to the formation of thick polymer coats that conceal the previously encountered negative charges over the surface of the uncoated cubosomes. The negative charges over the surface-coated particles might be attributed to the anionic nature of Eudragit® L100-55.
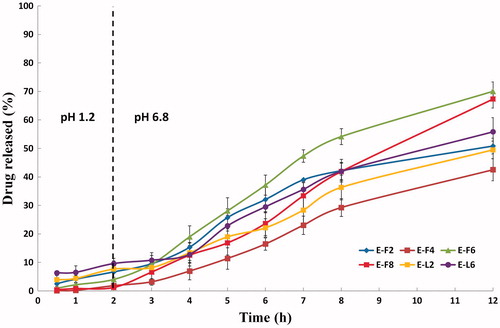
The in vitro release profiles of the selected PVS-loaded enteric surface-coated cubosomal dispersions in 0.1N HCl (pH 1.2, 2 h) and in Sorensen’s phosphate buffer (pH 6.8, 10 h) at 37 ± 0.5 °C displayed the delayed release characteristics of enteric-coated systems, . The results demonstrated the pH dependent solubility and gastroretentive potential of Eudragit® L100-55. As per USP (Citation2013) specifications for enteric-coated formulations, all systems showed P2h percentages lower than 10%. Furthermore, they developed sustained drug release profiles, for at least 12 h, at pH 6.8 (. Sharma et al. (Citation2011) correlated these findings with the protonation or deprotonation of Eudragit® L100-55 at various pH values. The carboxylic groups of Eudragit® L100-55 are protonated at pH values below the pKa of methacrylic acid of 4.23. Under the simulated acidic milieu of the stomach (pH 1.2), the enteric surface-coated nanocubosomes were protonated. This is expected to decrease the surface charges and the electrostatic repulsion between particles. This is usually accompanied by an increase the Vander Waals forces and the aggregation tendency of the coated cubosomes. In the simulated small intestine milieu (pH 6.8), the reverse occurs and the carboxylic groups are deprotonated. This is expected to restore the negative charges of the particles and the deaggregation tendency of the coated particles (Tayel et al., Citation2015). Needless to say, the observed drug release profiles at pH 6.8 showed comparable trends to those achieved with the uncoated systems; proving the impact of GMO: PVS ratio and the surfactant type on the drug released percentages.
Figure 6. In vitro release profiles of the selected PVS-loaded enteric surface-coated cubosomal dispersions in 0.1N HCl (pH 1.2, 2 h) and in Sorensen’s phosphate buffer (pH 6.8, 10 h) at 37 ± 0.5 °C (mean ± S.D., n = 3).
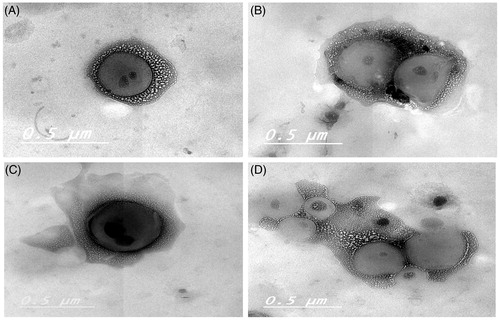
TEM micrographs of two representative enteric surface-coated dispersions (E-F8 and E-L6) were graphically illustrated in . It was revealed that the surface-coating technique had altered the morphology of the uncoated cubosomes. The cubosomes of the former dispersion (E-F8) were prepared by the fragmentation method and could be seen as more or less cubic nanoparticles (). On contrary, the liquid precursor-derived cubosomes were larger and showed structures with more circular edges () (Spicer & Hayden, Citation2001; Esposito et al., Citation2003; Rizwan et al., Citation2011). The appearance of multiple black dots within cubosomes was previously attributed to the presence of internal water channels (Madheswaran et al., Citation2014). Most of the coated cubosomes were more or less spherical in shape. However, the presence of cubic and/or irregular shaped particles was revealed; confirming the formation of other types of particles along with the targeted surface-coated cubosomes.
Figure 7. TEM micrographs of representative enteric surface-coated cubosomal dispersions E-F8 (A, B) and E-L6 (C, D).
In vivo absorption studies in rats
PVS pharmacokinetics following oral administration of an aqueous drug solution (treatment A) and the best achieved enteric surface-coated cubosomal dispersion (E-F8, treatment B) was evaluated in rats. The plasma concentration – time curves of the treatments clarified the rapid drug absorption characteristics of the former and the delayed drug release pattern of the latter, .
Figure 8. Plasma concentration-time curves of PVS-loaded enteric surface-coated cubosomal dispersion (E-F8) and an aqueous drug solution following oral administration in rats (mean ± S.D., n = 5).
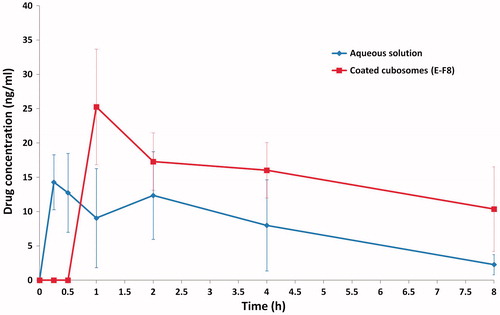
PVS solution was rapidly absorbed and showed two absorption peaks of 14.28 ng/ml and 12.35 ng/ml at 0.25 h and 2 h, post dosing. The maximum drug concentration (Cmax) of 15.61 ± 4.92 ng/ml was achieved at a Tmax of 0.350 ± 0.136 h (Tayel et al., Citation2015). The double-peak phenomenon in the drug plasma concentration–time curves was previously correlated to the rapid drug absorption at its main site of absorption and the delayed drug re-absorption from the enterohepatic circulation into the circulation, respectively (Sun et al., Citation2009; Shirasaka et al., Citation2010). Needless to say, PVS is one of the drugs that undergo extensive first pass metabolism where around 50% of dose is metabolized before reaching the systemic circulation (Quion & Jones, Citation1994).
Two theories were proposed to describe the rapid absorption characteristics of PVS from the duodenum. The first proposal is based on the pH-partition hypothesis that suggests that PVS is absorbed via passive diffusion. In fact, the validity of this proposal could not be guaranteed because of the hydrophilic nature of PVS (log p value of - 0.84) that should limit its penetration across membranes via passive diffusion only (Shitara & Sugiyama, Citation2006). The more relevant proposal is based on presence of specific transporting carriers for PVS in the small intestine. At the acidic microclimate pH around the intestinal epithelial cells, PVS molecules exist as anions. The membrane transport of this hydrophilic statin is probably mediated by the organic anion transporting polypeptides (OATPs; mainly Oatp1a5) expressed in the small intestine (Sun et al., Citation2009).
One absorption peak with a Cmax of 25.88 ± 7.09 ng/ml was observed following the oral administration of the coated cubosomal dispersion (E-F8) in rats. The ability of the coating polymer (Eudragit® L 100-55) to protect the entrapped PVS from its possible degradation in the stomach (to 3′α-isopravastatin and 6′-epipravastatin) and the subsequent delivery to the duodenum was evidenced by the significant (p < 0.01) delay in the mean Tmax from 0.35 h (treatment A) to 1.20 h (treatment B). Aqueous solutions and dispersions are rapidly emptied from the stomachs of the fasted rats within 15 – 30 min (Nilsson & Johansson, Citation1973; Kendall et al., Citation2009). The pH of the duodenum acts as a trigger to promote the dissolution of the enteric coat and to allow the delivery of PVS-loaded cubosomes to its main site of absorption. Several factors could account for the significantly (p < 0.01) higher Cmax value of this treatment, including; the amorphous nature of the loaded drug, the large surface area to volume ratio offered by cubosomes as well as the presence of a certain percentage of the un-entrapped drug within the dispersions.
The significantly (p < 0.001) prolonged MRT(0−∞) (11.64 h vs 3.86 h) for treatment B and treatment A, respectively could indicate the controlled-release characteristics of the developed enteric surface-coated system.
The increase in the oral PVS bioavailability was found to be 3.28 folds; with respect to the estimated mean AUC(0−∞) values of treatment A (70.140 ng.h/ml) and treatment B (230.650 ng.h/ml). The higher oral bioavailability could be related to the ability of the enteric surface-coated cubosomes to protect PVS from the hostile gastric environment; as evidenced from the hindered drug absorption in the first 30 min, to target PVS to its main site of absorption at the duodenum; as proved by the significantly higher Cmax values and to minimize the significant first pass metabolism; as deduced from the disappearance of the second absorption peak.
Conclusions
The enteric surface-coating of the pre-fabricated nanocubosomal dispersions was successfully achieved, in a single step and without the need of organic solvents, via the freeze drying technique. The current work demonstrated the ability of the developed systems to trigger the release of PVS at its main site of absorption in the duodenum and subsequently, to sustain the absorption of this freely water soluble drug along the small intestine in rats. A more comprehensive research in this direction is required to evaluate the possible intrinsic potential of these enteric surface-coated systems for other hydrophilic drugs.
Declaration of interest
The authors report no declaration of interest.
References
- Bejugam NK, Uddin AN, Gayakwad SG, D'Souza MJ. (2008). Formulation and evaluation of albumin microspheres and its enteric coating using a spray-dryer. J Microencapsul 25:577–83
- Chandy T, Rao GH, Wilson RF, Das GS. (2002). Delivery of LMW heparin via surface coated chitosan/peg-alginate microspheres prevents thrombosis. Drug Deliv 9:87–96
- Chong JY, Mulet X, Waddington LJ, et al. (2012). High-throughput discovery of novel steric stabilizers for cubic lyotropic liquid crystal nanoparticle dispersions. Langmuir 28:9223–32
- Clogston J, Rathman J, Tomasko D, et al. (2000). Phase behavior of a monoacylglycerol (Myverol 18-99K): water system. Chem Phys Lipids 107:191–220
- Eerikäinen H, Kauppinen EI. (2003). Preparation of polymeric nanoparticles containing corticosteroid by a novel aerosol flow reactor method. Int J Pharm 263:69–83
- Esposito E, Cortesi R, Drechsler M, et al. (2005). Cubosome dispersions as delivery systems for percutaneous administration of indomethacin. Pharm Res 22:2163–73
- Esposito E, Eblovi N, Rasi S, et al. (2003). Lipid-based supramolecular systems for topical application: a preformulatory study. AAPS PharmSci 5:E30
- Ganem-Quintanar A, Quintanar-Guerrero D, Buri P. (2000). Monoolein: a review of the pharmaceutical applications. Drug Dev Ind Pharm 26:809–20
- Ghorab DM, Amin MM, Khowessah OM, Tadros MI. (2011). Colon-targeted celecoxib-loaded Eudragit® S100-coated poly-ɛ-caprolactone microparticles: preparation, characterization and in vivo evaluation in rats. Drug Deliv 18:523–35
- Hao S, Wang B, Wang Y, et al. (2013). Preparation of Eudragit L 100-55 enteric nanoparticles by a novel emulsion diffusion method. Colloids Surf B Biointerfaces 108:127–33
- Hatanaka T. (2000). Clinical pharmacokinetics of pravastatin: mechanisms of pharmacokinetic events. Clin Pharmacokinet 39:397–412
- Kendall RA, Alhnan MA, Nilkumhang S, et al. (2009). Fabrication and in vivo evaluation of highly pH-responsive acrylic microparticles for targeted gastrointestinal delivery. Eur J Pharm Sci 37:284–90
- Kivistö KT, Grisk O, Hofmann U, et al. (2005). Disposition of oral and intravenous pravastatin in MRP2-deficient TR- rats. Drug Metab Dispos 33:1593–6
- Kojarunchitt T, Hook S, Rizwan S, et al. (2011). Development and characterisation of modified poloxamer 407 thermoresponsive depot systems containing cubosomes. Int J Pharm 408:20–6
- Komai T, Kawai K, Tokui T, et al. (1992). Disposition and metabolism of pravastatin sodium in rats, dogs and monkeys. Eur J Drug Metab Pharmacokinet 17:103–13
- Kwon TK, Hong SK, Kim JC. (2012). In vitro skin permeation of cubosomes containing triclosan. J Ind Eng Chem 18:563–7
- Lee KW, Nguyen TH, Hanley T, Boyd BJ. (2009). Nanostructure of liquid crystalline matrix determines in vitro sustained release and in vivo oral absorption kinetics for hydrophilic model drugs. Int J Pharm 365:190–9
- Leroux JC, Cozens R, Roesel JL, et al. (1995). Pharmacokinetics of a novel HIV-1 protease inhibitor incorporated into biodegradable or enteric nanoparticles following intravenous and oral administration to mice. J Pharm Sci 84:1387–91
- Ma YR, Zhou Y, Zhang GQ, et al. (2014). Simultaneous determination of repaglinide and pravastatin sodium in rat plasma by LC-Ms/MS and its application on pharmacokinetic interactions study. Acta Pharm Sin B 49:72–7
- Madheswaran T, Baskaran R, Yong CS, Yoo BK. (2014). Enhanced topical delivery of finasteride using glyceryl monooleate-based liquid crystalline nanoparticles stabilized by cremophor surfactants. AAPS PharmSciTech 15:44–51
- Maghsoodi M. (2009). Physicomechanical properties of naproxen-loaded microparticles prepared from Eudragit L100. AAPS PharmSciTech 10:120–8
- Mariani P, Luzzati V, Delacroix H. (1988). Cubic phases of lipid-containing systems. Structure analysis and biological implications. J Mol Biol 204:165–89
- Martinho N, Damgé C, Reis CP. (2011). Recent advances in drug delivery systems. J Biomater. Nanobiotech 2:510–26
- Moebus K, Siepmann J, Bodmeier R. (2012). Cubic phase-forming dry powders for controlled drug delivery on mucosal surfaces. J Control Release 157:206–15
- Morsi NM, Abdelbary GA, Ahmed MA. (2014). Silver sulfadiazine based cubosome hydrogels for topical treatment of burns: development and in vitro/in vivo characterization. Eur J Pharm Biopharm 86:178–89
- Niazi SK. 2014. Handbook of bioequivalence testing. Appendix B: dissolution testing requirements for U.S. FDA submission. 2nd ed. London, England: CRC Press, 863
- Nigović B, Vegar I. (2008). Capillary electrophoresis determination of pravastatin and separation of its degradation products. Croatica Chemica Acta 81:615–22
- Nilsson F, Johansson H. (1973). A double isotope technique for the evaluation of drug action on gastric evacuation and small bowel propulsion studied in the rat. Gut 14:475–7
- Quion JAV, Jones PH. (1994). Clinical pharmacokinetics of pravastatin. Clin Pharmacokinet 27:94–103
- Rizwan SB, Assmus D, Boehnke A, et al. (2011). Preparation of phytantriol cubosomes by solvent precursor dilution for the delivery of protein vaccines. Eur J Pharm Biopharm 79:15–22
- Salunkhe SS, Bhatia NM, Bhatia MS. (2015). Implications of formulation design on lipid-based nanostructured carrier system for drug delivery to brain. Drug Deliv. [Epub ahead of print]. doi:10.3109/10717544.2014.943337
- Sastry SV, Nyshadham JR, Fix JA. (2000). Recent technological advances in oral drug delivery - a review. Pharm Sci Technol Today 3:138–45
- Sharma M, Sharma V, Panda AK, Majumdar DK. (2011). Development of enteric submicron particle formulation of papain for oral delivery. Int J Nanomedicine 6:2097–3111
- Shirasaka Y, Suzuki K, Nakanishi T, Tamai I. (2010). Intestinal absorption of HMG-CoA reductase inhibitor pravastatin mediated by organic anion transporting polypeptide. Pharm Res 27:2141–9
- Shitara Y, Sugiyama Y. (2006). Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: drug-drug interactions and inter-individual differences in transporter and metabolic enzyme functions. Pharmacol Ther 112:71–105
- Siekmann B, Bunjes H, Koch MHJ, Westesen K. (2002). Preparation and structural investigations of colloidal dispersions prepared from cubic monoglyceride-water phases. Int J Pharm 244:33–43
- Singhvi S, Pan H, Morrison R, Willard D. (1990). Disposition of pravastatin sodium, a tissue-selective HMG-CoA reductase inhibitor, in healthy subjects. Br J Clin Pharmacol 29:239–43
- Spicer PT, Hayden KL. (2001). Novel process for producing cubic liquid crystalline nanoparticles (cubosomes). Langmuir 17:5748–56
- Sun Y, Dai J, Hu Z, et al. (2009). Oral bioavailability and brain penetration of (-)-stepholidine, a tetrahydroprotoberberine agonist at dopamine D(1) and antagonist at D(2) receptors, in rats. Br J Pharmacol 158:1302–12
- Tayel SA, El-Nabarawi MA, Tadros MI, Abd-Elsalam WH. (2015). Duodenum-triggered delivery of pravastatin sodium via enteric surface-coated nanovesicular spanlastic dispersions: development, characterization and pharmacokinetic assessments. Int J Pharm 483:77–88
- Tayel SA, El-Nabarawi MA, Tadros MI, Abd-Elsalam WH. (2013). Positively charged polymeric nanoparticle reservoirs of terbinafine hydrochloride: preclinical implications for controlled drug delivery in the aqueous humor of rabbits. AAPS PharmSciTech 14:782–93
- Talluri SV, Kuppusamy G, Reddy Karri VV, et al. (2015). Lipid-based nanocarriers for breast cancer treatment - comprehensive review. Drug Deliv. [Epub ahead of print]. doi:10.3109/10717544.2015.1092183
- United States Pharmacopeia and National Formulary USP 36–NF 31, 2013. (711) Dissolution. The United States Pharmacopeial Convention, Inc. Rockville, MD
- Wacker M. (2013). Nanocarriers for intravenous injection-the long hard road to the market. Int J Pharm 457:50–62
- Wörle G, Dreschler M, Koch MHJ, et al. (2007). Influence of composition and preparation parameters on the properties of aqueous monoolein dispersions. Int J Pharm 329:150–7
- Zhang S, Kawakami K, Yamamoto M, et al. (2011). Coaxial electrospray formulations for improving oral absorption of a poorly water-soluble drug. Mol Pharm 8:807–13