
Abstract
It was the first time that 3-octadecylcarbamoylacrylicacid-cisplatin nanocomplexes (OMI-CDDP-N) were synthesized via monocarboxylato and O→Pt coordination. The nanocomplexes exhibited lower IC50 values compared with cisplatin (CDDP) in vitro, and enhanced antitumor efficacy in murine liver cancer (H22) in vivo. However, the toxicity of the nanocomplexes was more severe than CDDP. OMI-CDDP-N-based liposome (OCP-L) was prepared in order to maintain the efficacy and reduce the toxicity of OMI-CDDP-N. Here, a series of parameters were investigated to optimize liposome formulation. The optimal formulation was CDDP/phospholipids/cholesterol (1/10/0.7 wt.%), with distilled water as hydration medium and an encapsulation efficiency of 94.2 ± 2.1%. In vitro cytotoxicity studies revealed that OCP-L and OMI-CDDP-N exhibited a lower IC50 compared with commercial cisplatin injection (CDDP-S) in a variety of human cancer cells. In H22-implanted mice, OCP-L showed a significantly higher antitumor activity than OMI-CDDP-N or CDDP-S at a dose of 5 mg/kg (p < 0.01). Moreover, OCP-L exceeded the size cutoff for kidney clearance, hence it bypassed the nephrotoxicity of CDDP which is a major curse of CDDP in the clinic. These results suggested that the unique OMI-CDDP-N-entrapped liposome can overcome the disadvantages associated with conventional CDDP therapy and provide a higher safety profile.
Introduction
Cisplatin (CDDP) is one of the most widely used and effective chemotherapeutic agents for the treatment of testicular, ovarian cancers, bladder and small cell lung cancer (Rosenberg & Vancamp, Citation1969; Fricker, Citation2007; Wheate et al., Citation2010). However, its therapeutic efficacy has been adversely affected by its side effects, mostly nephrotoxicity, ototoxicity and neurotoxicity (Boulikas et al., Citation2007). A widely accepted explanation of antitumor activity of CDDP is based on their aquated form binding to DNA fragments, generally adjacent guanosines (G), with subsequent distortion of DNA replication and transcription via producing intrastrand N7(G)-Pt-N7(G) kink structure (Boulikas & Vougiouka, Citation2003; Kasparkova et al., Citation2008). Therefore, aquation is one of the key processes in the pharmacology of CDDP. In aqueous media, CDDP undergoes step-wise aquation reactions, in which the chloride ions are replaced by water molecules resulting in the formation of cationic aquated form. The rates of aquation of CDDP are mainly determined by the concentration of the chloride ions in biological fluids. After administration, CDDP experiences a high chloride concentration (100 mM) that prevents its hydrolysis to the aquated form. In contrast, the low concentration of chloride concentration in the cells (4–12 mM) favors the formation of cationic aquated form. Furthermore, the rate of exchange of chloride ligands with water molecules affects the efficacy and toxicity of CDDP. Rapid aquation in the blood produces highly active platinum species which react with various molecules in the blood and cause severe systemic toxicity, while slower aquation could reduce toxicity and prolong the plasma half-life, unfortunately, reduce the antitumor activity of the drug (Knox et al., Citation1986; Johnstone et al., Citation2013; Oberoi et al., Citation2013; Shaili, Citation2014).
In an effort to improve the efficacy and reduce the adverse effects of CDDP, nanoparticle CDDP formulations appear to be very promising. Nanoparticulate CDDP formulations have been achieved by chelating CDDP with polymers or encapsulating CDDP into liposomes (Xiong et al., Citation2012; Guo et al., Citation2013; Li et al., Citation2014; Wang et al., Citation2014). For example, Uchino et al. (Citation2005) chelated CDDP positively charged platinum species to carboxylate-rich copolymers with a drug loading of 30 wt.%. Significantly antitumor activity was observed on the murine colon cancer (C26)-implanted mice model, while blood biochemistry revealed transient hepatotoxicity. SPI-077, a liposomal CDDP, exhibited a 28-fold higher tumor exposure to platinum with a four-fold lower platinum exposure to kidneys relative to CDDP-treated animals. However, the clinical antitumor efficacy was modest, which resulted in early closure of the trial (Kim et al., Citation2001; Vail et al., Citation2002). Recently, Abhimanyu S. Paraskar et al. found that the complexation environment of the platinum had an impact on the efficacy of CDDP. The monocarboxylato and O→Pt coordination environment between the platinum and the leaving group were more efficient in releasing CDDP in a pH-dependent manner than coordination via more stable dicarboxylato linkages (Paraskar, et al., Citation2010; Sengupta et al., Citation2012).
In this paper, we have developed 3-octadecylcarbamoylacrylic acid-CDDP nanocomplexes (OMI-CDDP-N) through monocarboxylato and O→Pt coordination to increase the therapeutic efficacy of CDDP. An optimal number of OMI/CDDP molar ratios were selected to maintain good stability. Such optimized OMI-CDDP-N was incorporated into the liposomes (OCP-L) to reach superior anticancer therapeutics along with decreased renal toxicity. The production of liposomes will be investigated under different operative conditions: the ratio of phospholipids/cholesterol, the type of phospholipids, hydration medium and the ratio of CDDP/phospholipids. In addition, the comparative studies of in vitro cytotoxicity, in vivo anticancer efficacy and safety of commercial CDDP injection (CDDP-S), OMI-CDDP-N and OCP-L were carried out.
Materials and methods
Materials
N-octadecylamine and maleic anhydride were obtained from Sigma-Aldrich (St. Louis, MO). CDDP was obtained from Shandong Boyuan Chemical Co., Ltd. (Shandong, China). Soybean phospholipids (SPC) were obtained from Lipoid Co., Ltd. (Ludwigshafen, Germany). CDDP-S was obtained from Qilu Pharmaceutical Co., Ltd. (Shandong, China). All other reagents were of analytical grade and used as received.
Synthesis of OMI-CDDP-N
OMI was synthesized according to the previously reported method (Mallik et al., Citation2010). A solution of N-octadecylamine (0.408 g, 1.51 mmol) in chloroform (25 mL) was added dropwise to a solution of maleic anhydride (0.630 g, 6.42 mmol) in chloroform (25 mL), and the mixture was stirred for 30 min at room temperature. The obtained solid was vacuum filtered and washed with chloroform (100 mL) and acetone (100 mL) to obtain OMI as a white solid (90% yield). OMI was characterized by FT-IR spectrum (VERTEX 70V FT-IR, Bruker, Germany) at 25 °C.
For synthesis of OMI-CDDP-N, the various ratios of OMI and CDDP were dissolved in distilled water and allowed to react at 55 °C with gentle stirring to form the OMI-CDDP-N. The nanocomplexes were characterized by FT-IR spectrum (VERTEX 70V FT-IR, Bruker, Germany) at 25 °C.
Preparation of OMI-CDDP-N based liposomes
Reverse phase evaporation (REV) was used to prepare OCP-L. Briefly, the lipids were dissolved in diethyl ether and then an aqueous solution containing the OMI-CDDP-N was added to the lipid mixture to form a W/O emulsion. The emulsions were sonicated for approximately 10 min, then the sonication vessel was transferred directly to a rotary evaporator and the contents were dried down gently at 40 °C until a gel was formed. The volume of the resulting gel was then adjusted to 6 mL with an aqueous solution. After the suspension was formed, drying was continued for another 20 min on the evaporator at 40 °C to remove any traces of the solvent. The resulting liposomes were sterilized by passage through a 0.22 μm filter. Various factors comprising the ratio of SPC/cholesterol, the type of phospholipids, the hydration medium and the ratio of CDDP/SPC were investigated to find the optimum condition for liposome formation.
Characterization of OMI-CDDP-N and OCP-L
The morphologies of OMI-CDDP-N and OCP-L were observed using a JEOL-1210 transmission electron microscope (TEM) (JEOL, Tokyo, Japan) at 200 kV. The OMI-CDDP-N and OCP-L were diluted 20-fold with distilled water, of which the particle size, distribution and zeta potential were determined using a Zetasizer (Nano ZS, Malvern Instruments, Malvern, UK).
Encapsulation efficiency and drug loading capacity
Encapsulation efficiency (EE) was determined using a Model 8010 ultrafiltration device (Millipore Corporation, Bedford, CA) with a molecular weight cutoff of 50 kDa. Briefly, 0.5 mL of the OCP-L was diluted 10-fold with distilled water. The free CDDP (Cout) was separated by ultrafiltration through a Millipore filter and assayed using the validated derivatization High-Performance Liquid Chromatography (HPLC) method (Andrews et al., Citation1984). The total drug (C) was determined after the liposome suspension was diluted with 400 μL pepsase (2%) to disrupt the liposomes completely and release the encapsulated drug into the solvent.
The EE% was calculated according to the following Equation:
Drug-loading capacity (LC) of OCP-L was expressed by the amount of encapsulated drug (WE) to the amount of total lipids (Wp) mass ratios:
Release kinetics of CDDP from OMI-CDDP-N and OCP-L
In vitro drug release studies were carried out using a dialysis method, tubing cellulose membrane with an average flat width of 25 mm and 10 kDa cut off (Millipore Corporation, Bedford, CA). The prepared OMI-CDDP-N and OCP-L were suspended in PBS buffer (pH = 7.4) and transferred to the dialysis tubes. Then, this system was subjected to dialysis by submerging the tubing in a breaker containing 200 mL buffer. The dialysis processes were carried out using a magnetic stirrer at 37 °C. At suitable intervals, 5 mL samples were removed and replaced with fresh buffer to ensure a constant volume. The sample was determined using the validated derivatization-HPLC method.
Animals and tumor cell lines
KM mice (male, 18–22 g, a total of 70) were obtained from the Laboratory Animal Center of Shenyang Pharmaceutical University (Shenyang, China). All animal experiments were carried out in accordance with the Institutional Animal Care and Use Committee of Shenyang Pharmaceutical University.
Streptomycin–penicillin solution, 0.25% Trypsin-EDTA, human breast cancer cells (MCF-7), human liver cancer cells (HEPG-2), human lung cancer cells (A549) and murine liver cancer cells (H22) were from Nanjing KeyGEN Biotech. Co., Ltd. (Nanjing, China). Fetal bovine serum (FBS) was obtained from ExCell Biology (Shanghai, China). DMEM and RPMI 1640 were obtained from Gibco Inc. (Invitrogen, Carlsbad, CA). HEPG-2 and MCF-7 were grown in a DMEM media. A549 and H22 cells were grown in RPMI 1640 media. All media were supplemented with 10% FBS and 1% penicillin–streptomycin solution. All cells were grown at 37 °C with 5% CO2.
In vitro cytotoxicity
In vitro cytotoxicity was assessed by MTT assay. MCF-7, HEPG-2 and A549 cancer cells were seeded in 96-well plates (1 × 105 cells/well) and grown at 37 °C. After 20 h, the medium was replaced with fresh media containing different concentrations (1–32 μM) of CDDP-S, OMI-CDDP-N and OCP-L. These plates were incubated at 37 °C. After 48 h, the medium was replaced with 100 μL fresh medium and the MTT reagent was added to each well and the plates were incubated for 3 h. The color intensity was measured at 570 nm using a microplate reader (Spectra Max M3, Molecular Devices, Sunnyvale, CA).
In vivo toxicity study
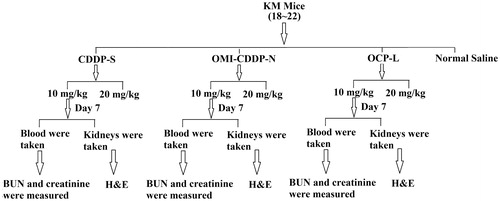
Male KM mice were randomly divided into seven groups (n = 6) and treated with normal saline, CDDP-S (10 mg/kg), CDDP-S (20 mg/kg), OMI-CDDP-N (10 mg/kg on CDDP basis), OMI-CDDP-N (20 mg/kg on CDDP basis), OCP-L (10 mg/kg on CDDP basis) and OCP-L (20 mg/kg on CDDP basis), respectively, by intravenously (i.v.) injection on day 0 (). The systemic toxicity was assessed by measuring the body weight and the mortality of the mice for 14 days. In the case of administering normal saline, CDDP-S, OMI-CDDP-N and OCP-L at a dose of 10 mg/kg on a CDDP basis, samples of blood and kidneys were taken on day 7 after administration. In each blood sample, plasma concentrations of blood urea nitrogen (BUN) and creatinine were measured. The kidneys were fixed in 10% formalin and blocks were paraffin embedded, sectioned, and stained with H&E using standard methods (Mizutani et al., Citation2002).
Figure 1. Study flow-chart in vivo toxicity.
In vivo antitumor efficacy
Tumor-bearing mice were established by subcutaneous inoculation of H22 liver cancer cells (1.6 × 106) into the right flank of male KM mice. After the tumor volume reached approximately 100 mm3, the mice were randomly divided into four groups (n = 7), and given i.v. injections of normal saline, CDDP-S, OMI-CDDP-N and OCP-L at an equivalent CDDP dose of 5.0 mg/kg twice at a 3-day interval. Tumor growth and body weight were monitored. Tumor volume was calculated using the following formula:
where the tumor length was L and the width measured by electronic calipers was W.
Statistical analysis
All results were reported as mean ± standard deviation (SD). The statistical significance of differences was evaluated by using a 2-sample t-test with a significance level of 0.05.
Results and discussion
Synthesis of OMI-CDDP-N
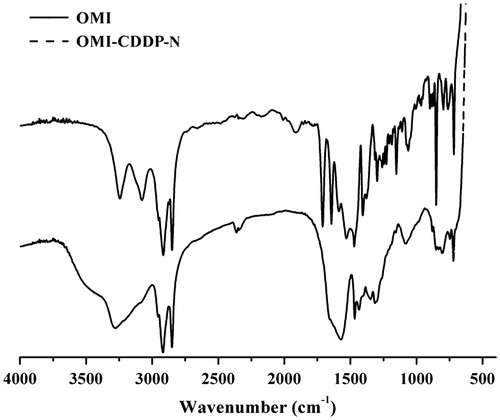
As outlined in , a coordination bond between the platinum (II) atom in CDDP and the carboxyl group in the side chain of OMI resulted in the spontaneous formation of OMI-CDDP-N. The nanocomplexes were indicated by the FT-IR spectroscopy (). The –NH bending vibrations at 1587 and 1527 cm−1 in OMI was shifted toward 1469 and 1433 cm−1 in nanocomplexes. Additionally, in nanocomplexes, there was a shift in –CO– stretching of –CONH– from 1643 to 1571 cm−1. The most significant confirmation of conjugate formation was the disappearance of the alcoholic hydroxyl group (3076 cm−1, 920 cm−1) from OMI.
Figure 2. Scheme for the synthesis of OMI-CDDP-N. Schematic representation shows the preparation of OMI-CDDP-N by self-assembly from OMI and CDDP.
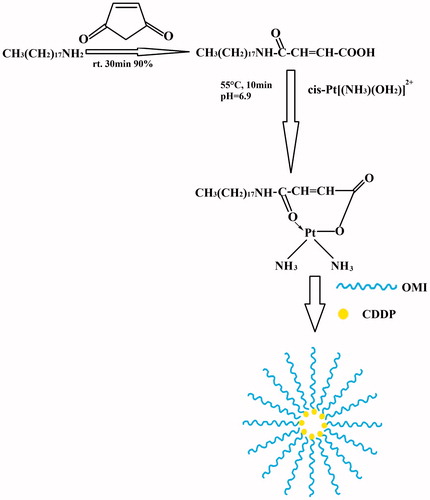
Figure 3. FT-IR spectra of OMI (upper) and OMI-CDDP-N (lower).
In this paper, synthesized OMI-CDDP-N appeared as a white dispersion in contrast to the yellow CDDP. When the molar ratio of the OMI/CDDP was 0.5, yellow CDDP precipitates were observed. While at ratios of 1/1 to 2/1, there was no CDDP precipitate. Uniform white dispersion was observed on 1/1 and 3/2 ratios, while aggregates of complexes were found at a higher ratio. Therefore, OMI/CDDP ratio of 1/1 was used for further optimization of liposome.
Factors influencing EE of OCP-L
Effect of the cholesterol content
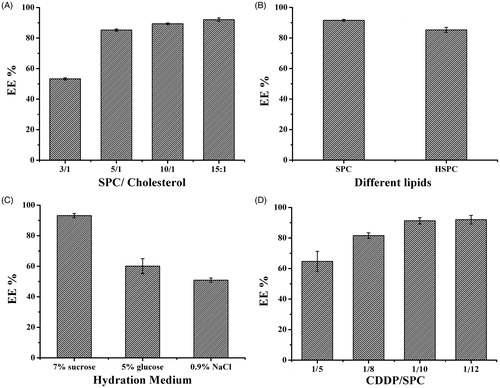
The effect of incorporating cholesterol within the lipid composition on EE was determined by varying the cholesterol contents (). The presence of cholesterol within the liposome bilayer had an effect on OMI-CDDP-N incorporation. An increase in EE% of OCP-L was observed with the ratio of SPC/cholesterol from 3/1 to 15/1 and the highest value of EE% was 92.1 ± 1.1% by 15/1. It was suggested that, the major reduction in drug incorporation when cholesterol content was further increased may be due to two conflicting factors: with increasing cholesterol, the bilayer hydrophobic and stability increased (Gregoriadis & Davis, Citation1979; Bernsdorff et al., Citation1997) and the permeability decreased (Kirby et al., 1980) which led to efficiently trapping the drug into bilayer as vesicles formed. In contrast, the long-chain carboxylate complexes of OMI-CDDP-N and cholesterol would both incorporate their carbon chains into the liposome bilayer. Higher amounts of cholesterol may compete with OMI-CDDP-N for packing space within the bilayer when the drug input was equal to or higher than the encapsulation capacity. Another study suggested that increasing cholesterol beyond a certain concentration could disrupt the regular linear structure of vesicular membranes, which decreased the EE of OMI-CDDP-N.
Figure 4. Encapsulation efficiency of OCP-L prepared by various conditions. (A) Encapsulation efficiency of CDDP in OCP-L prepared at various ratios of SPC/cholesterol. (B) Encapsulation efficiency of CDDP in OCP-L prepared by different types of phospholipids. (C) Encapsulation efficiency of CDDP in OCP-L prepared at different hydration medium. (D) Encapsulation efficiency of CDDP in OCP-L prepared at different ratios of CDDP/SPC.
Effect of the type of phospholipids
The EE of OMI-CDDP-N in liposome depended on the type of phospholipids used for the preparation of the lipid bilayer. As depicted in , the higher OMI-CDDP-N encapsulation was founded in SPC liposome with the value of 91.6 ± 0.6% while the lower loading was observed in HSPC formulation (85.3 ± 1.7%). This increase in percent encapsulation of SPC can be explained that the OMI-CDDP-N tended to be incorporated more efficiently in fluid membranes where the fatty acyl side chains had considerable freedom of movement. The introduction of double bonds into acyl chains increased the fluidity of lipid membrane, which, consequently, raised the lipid barrier permeability. These appealing results are in agreement with the fact that increasing mobility of the chains in the lipid membrane greatly enhances the EE of hydrophobic drugs (Peetla et al., 2009).
Selection of hydration medium
The EE of liposome with different hydration medium is shown in , which revealed that different hydration medium imposed strong influence on the EE of OMI-CDDP-N based liposome. The maximum EE was 93.2 ± 1.4% when the hydration medium was distilled water. However, the decrease in EE was significant when the hydration mediums were 5% glucose or 0.9% NaCl. Since aquated species of CDDP will undergo ligand exchange reaction with carboxylate ligands in neutral pH, and the nanocomplexes would exhibit dissociation at pH 3.5 using 5% glucose, it resulted in a tremendous reduction in EE (only 60.1 ± 4.9%). These unfavorable properties of aquated species would limit the EE and cause severe systemic toxicity. Addition of Cl- into the hydration medium seemed to have significant influence on the EE of liposomes as shown in . The Cl− apparently resulted in a decline of EE of liposomes. The possible reason was that OMI-CDDP-N would slowly release free CDDP in the presence of abundant chloride ions due to the coordination bonds between the atoms of Pt of CDDP and the carboxylic group in the side chain of OMI. These may help to explain the occurrence of large loss of CDDP into the outer phase.
Effect of the CDDP/SPC mass ratio
In addition to promoting biocompatibility and acting as a physical barrier that minimizes drug leakage from the liposome, the SPC lipid layer also functions as a surfactant stabilizer of the W/O emulsions. Therefore, the optimal amount of SPC required to prepare the liposome must be determined. The effect of the CDDP/SPC mass ratio on the EE of OCP-L is shown in . The EE of OCP-L increased at the CDDP/SPC mass ratios from 1/5 to 1/10. However, no significant increase in EE was observed even when the CDDP/SPC mass ratio was increased up to 1/12 (92.0 ± 2.8%). Being a low water-soluble agent, CDDP was not suitable for entraping in the compartment of liposome. This in turn also required a very high lipid composition. High total lipid contents used in preparation would result in more lipid-carrier formation, which explained the corresponding increase in the encapsulation yield (Gregoriadis & Davis, Citation1979).
Characterization of OMI-CDDP-N and OCP-L
The ultrastructure analysis using TEM revealed that OMI-CDDP-N and OCP-L were spherical in aqueous solution (). Visual determination of OMI-CDDP-N and OCP-L size was accomplished using dynamic light scattering, showing that OMI-CDDP-N and OCP-L had an average size of 57.9 ± 1.1 nm and 110.7 ± 3.1 nm with a narrow distribution, suggesting that the nanocomplexes and liposome could potentially reduce systemic side effects, and exhibit increased intra-tumoral delivery by enhancing permeation and retention (EPR) effect (Maeda, Citation2001).
Figure 5. TEM images of OMI-CDDP-N and OCP-L. OMI-CDDP-N and OCP-L were negatively stained with phoshotungstic acid. Scale bar represents 200 nm.
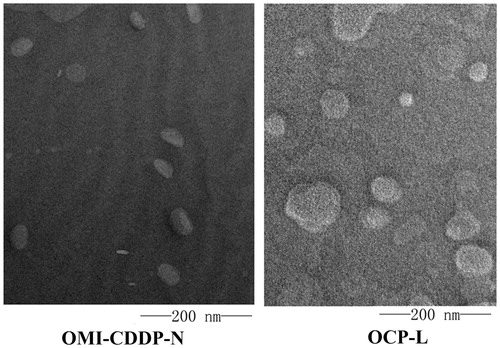
The surface charge of OMI-CDDP-N and OCP-L were −48.5 mV and −30.1 mV, as shown by zeta potential analysis. This negative zeta potential helps particles repel against each other in the suspension, ensuring long-term stability and avoiding particle aggregation (Nukolova et al., Citation2011).
Encapsulation efficiency and drug loading capacity
The EE% of OCP-L was found to be 94.2 ± 2.1% and the LC% of OCP-L was about 10.0 ± 1.2%, which were dependent on the content of OMI in the liposomes. Most conventional liposomal CDDP formulations had low EE% and LC% due to its unfavorable properties, such as poor water solubility and low lipophilicity. Better EE would be achieved by increasing the lipid concentration. However, the complexation of aquated species with long-chain carboxylate ligands provided a high entrapment of CDDP along with reducing the content of lipid. The LC% of OCP-L was found to be 10.0 ± 1.2%, which was significantly higher than that of two clinically evaluated CDDP liposomes SPI-077 (6.7 wt.%) and lipoplatin (10 wt.%) (Vail et al., Citation2002; Jehn et al., Citation2007;).
In vitro drug release profile
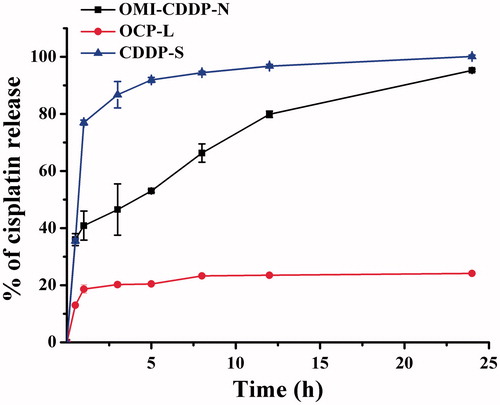
The in vitro drug release profiles of CDDP-S, OMI-CDDP-N and OCP-L over 24 h are shown in . The sustained release of CDDP from the OMI-CDDP-N and OCP-L was due to the presence of abundant chloride ions in the buffer. It can be seen that there was an initial burst release of 50.0 ± 0.5% for OMI-CDDP-N, 20.4 ± 0.3% for OCP-L, and 91.9 ± 0.9% for CDDP-S in the first 5 h. The initial burst of OCP-L happened probably due to the relatively high concentration of OMI-CDDP-N at the surface of the OCP-L. After 24 h, the cumulative release reached 24.1 ± 0.3% for OCP-L. The drug released from the OMI-CDDP-N was significantly faster than that of OCP-L. For example, up to 50% of the drug released from OMI-CDDP-N during first 5 h. The accelerated release of OMI-CDDP-N may be due to the release of free CDDP in the presence of abundant chloride ions, which weakens the coupling between the atoms of Pt (II) of CDDP and the carboxylic group in the side chain of OMI. Another factor contributing to a faster release was the large surface area due to the small particle size of OMI-CDDP-N. In addition, the OCP-L and OMI-CDDP-N both exhibited a significantly lower rate of CDDP release compared with CDDP-S with an initial burst of 91.9 ± 0.9%.
Figure 6. Graph shows the release of CDDP from OMI-CDDP-N and OCP-L as quantized over a 24 h periods (pH = 7.4 buffers). Data represent mean ± SD, n = 3.
In vitro cytotoxicity
All the CDDP drug formulations (CDDP-S, OMI-CDDP-N and OCP-L) investigated in this study were found to inhibit the growth of MCF-7, HEPG-2 and A549 in a concentration-dependent manner. As shown in , CDDP-S exhibited significantly less cytotoxicity compared with OMI-CDDP-N in terms of the growth inhibition in MCF-7, HEPG-2 and A549, respectively (half maximal inhibitory concentration IC50: 15.7 μM vs. 7.26 μM, p < 0.01, 6.55 μM vs. 3.28 μM, p < 0.01 and 8.16 μM vs. 6.18 μM, p < 0.05). As expected, OCP-L was the most effective among the three drug formulations. The higher cytotoxicity of OMI-CDDP-N and OCP-L might either be due to introduction of the monocarboxylato and O→Pt coordination environment which facilitates rapid activation of platinum and react with the nitrogen bases of DNA, or the direct ligand exchange reaction between the complex and DNA nitrogen bases. To be noted, OCP-L decreased the IC50 value compared to OMI-CDDP-N. It revealed the fact that OCP-L could enhance the drug accumulation in the cells due to perturbation effect on the cell membrane and reduce its permeability barrier. This effect could facilitate the uptake of anticancer drugs at the target cell membranes. Moreover, the OCP-L enabled enhanced intratumoral concentrations and efficient released active platinum moiety in a pH-dependent behavior (data not shown).
Table 1. IC50 values (μM) of CDDP-S, OMI-CDDP-N and OCP-L in three cancer cell lines.
In vivo toxicity study
The survival rates and percent change in body weight were measured for each animal and the results are shown in . The mice receiving OCP-L or normal saline at a dose of 10 mg/kg showed an increase in body weight without any observable side effects. However, mice in the CDDP-S and OMI-CDDP-N-treated group induced a significant reduction in body weight more than 20%. Moreover, the survival rates were 0% when mice received CDDP-S and OMI-CDDP-N at a dose of 20 mg/kg. Interestingly, the survival rates of OMI-CDDP-N were lower than that of CDDP-S at the same dose. The main reason was that the monocarboxylato and O→Pt coordination bond between the platinum (II) atom in CDDP and the carboxyl group in the side chain of OMI resulted in the rapid aquation, which caused severe systemic toxicity. In addition, the OMI-CDDP-N released their drug content at a rate that was rapid compared to the rate of OCP-L, which enhanced the systemic toxicity.
Figure 7. In vivo toxicity studies of CDDP-S, OMI-CDDP-N and OCP-L in normal mice. (A) Survival rates of mice treated with normal saline, CDDP-S, OMI-CDDP-N and OCP-L at a dose of 10 mg/kg and 20 mg/kg. (B) Changes in relative body weight of the mice treated with normal saline, CDDP-S and OCP-L at a dose of 10 mg/kg. Data are presented as the mean ± SD, n = 6. (C) Histological images from the kidney of the treated mice. Images were taken at × 40 magnifications with standard H&E staining.
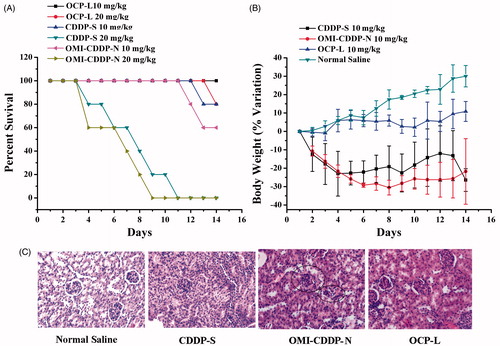
Kidney damage was assessed by analyzing creatinine, BUN and histopathology. As shown in , BUN levels in the normal saline and OCP-L 10 mg/kg treatment group were normal, (9.50 ± 1.45 mmol/L vs. 8.28 ± 1.29 mmol/L, respectively) (p > 0.05). The BUN levels in the CDDP-S 10 mg/kg treatment group were 14.46 ± 0.93 mmol/L, showing no difference compared with that in the OMI-CDDP-N 10 mg/kg treatment group (BUN: 16.78 ± 6.12 mmol/L, p > 0.05). The plasma concentrations of creatinine on day 7 after administration of CDDP-S, OMI-CDDP-N and OCP-L were 133.03 ± 27.77, 114.55 ± 17.72 and 69.31 ± 21.99 μmol/L, respectively (). The group given CDDP-S and OMI-CDDP-N 10 mg/kg showed significantly higher plasma concentrations of creatinine compared with the OCP-L 10 mg/kg group (p < 0.001). Histological analysis was used to confirm the nephrotoxicity of CDDP (). The histopathology criteria was assessed based on changes in tubular morphology, the presence of tubular dilation and protein casts, an indicator of extensive tubular damage. Normal renal morphology, determined by a clear lumen and an intact-brush border, was observed in the normal saline and OCP-L-treated animals. In contrast, light microscopy indicated the tubular dilation from all animals in the CDDP-S, OMI-CDDP-N treatment group. The low toxicity of OCP-L can be explained as follows: (1) The particle size of the OCP-L is approximately 110.7 ± 3.1 nm, it is now documented that nanoparticles larger than 5 nm will avoid renal clearance, and thereby could potentially reduce CDDP nephrotoxicity (Choi et al., Citation2007). (2) The toxicity of CDDP was related to peak plasma Pt concentration, more specifically, peak filterable Pt concentration (Nagai & Ogata, Citation1997). OCP-L avoids a high-peak plasma concentration of free, since most of the drug is retained in the liposome during circulation and gradually released with time. The tumor and other tissues, as well as plasma are continuously exposed to a low-dose CDDP environment, and the amount of filterable Pt remains proportionately low.
Table 2. The BUN levels of each group (n = 6).
Table 3. The creatinine levels of each group (n = 6).
In vivo anticancer efficacy
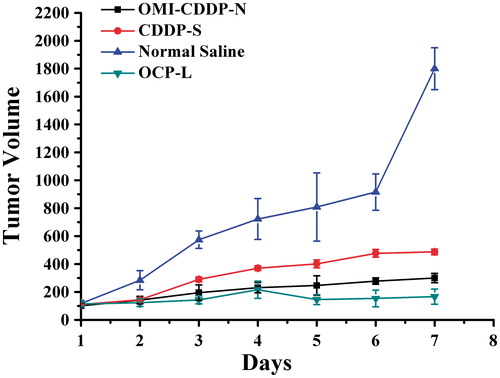
Efficacy of OMI-CDDP-N and OCP-L in a xenograft tumor model was further evaluated. As seen in , all CDDP treatments decreased tumor growth rates compared with the control vehicle (p < 0.05). Nevertheless, OCP-L treatment resulted in the greatest tumor growth inhibition (p < 0.01). The OMI-CDDP-N decreased the anti-tumor effect compared with OCP-L, probably due to the small size of OCP-L, which facilitated the accumulation of OCP-L in tumor cells through the EPR effect. Moreover, the sustained release of OCP-L would prevent the total CDDP release in the bloodstream and promote its release in endosomes or lysosomes in cancer cells. However, the CDDP-S only had a partial effect on tumor growth inhibition at the dose of 5 mg/kg in the experiment. Perhaps, the most important challenges addressed in this study were the delivery of the OMI-CDDP-N to an in vivo tumor and demonstration of a superior antitumor activity of the OCP-L. Surprisingly, similar to the in vitro experiments, the in vivo tumor delivery and anti-tumor effect were reduced by the OMI-CDDP-N.
Figure 8. Changes in the tumor volume of the H22 murine liver cancer tumor-bearing mice receiving intravenous injection of normal saline, CDDP-S, OMI-CDDP-N and OCP-L at equivalent CDDP dose of 5.0 mg/kg.
Conclusion
In this study, OMI-CDDP-N was prepared through monocarboxylato and O→Pt coordination. These nanoparticles were well-dispersed and stable in the aqueous solution. When CDDP was loaded in the OMI-CDDP-N, the size of OMI-CDDP-N was only 57.9 ± 1.1 nm. By adjusting the fabrication parameters, OMI-CDDP-N was readily incorporated into liposome by reverse phase evaporation approach with high-encapsulation efficiency (approximately 94.2%) and released CDDP in a sustained manner. In vitro cytotoxicity revealed that OCP-L had higher cytotoxicity compared to OMI-CDDP-N and CDDP-S against MCF-7, HEPG-2 and A549 cell lines. In vivo assay using normal KM mice treated with OCP-L had increased efficacy and decreased side effects, which was not obtained in the treatment with OMI-CDDP-N and CDDP-S. As platinum-based drugs become the frontline therapy for abroad spectrum of cancers, OCP-L with increased efficacy and reduced nephrotoxicity could probably be translated into the next-generation of nano-platinum drugs in the clinics.
Declaration of interest
The authors report no conflicts of interest. This work was supported by the Project of Innovation Team of University from the Education Department of Liaoning, China (LT201023) and the Beijing Sinuoer Biotech. Co., Ltd, China.
References
- Andrews PA, Wung WE, Howell SB, et al. (1984). A high-performance liquid chromatographic assay with improved selectivity for cisplatin and active platinum (II) complexes in plasma ultrafiltrate. Anal Biochem 143:46–56.
- Bernsdorff C, Wolf A, Winter R, et al. (1997). Effect of hydrostatic pressure on water penetration and rotational dynamics in phospholipid-cholesterol bilayers. Biophys J 72:1264.
- Boulikas T, Pantos A, Bellis E, et al. (2007). Designing platinum compounds in cancer: structures and mechanisms. Cancer Ther 5:537–83.
- Boulikas T, Vougiouka M. (2003). Cisplatin and platinum drugs at the molecular level (review). Oncol Rep 10:1663–82.
- Choi HS, Liu W, Misra P, et al. (2007). Renal clearance of quantum dots. Nat Biotechnol 25:1165–70.
- Fricker SP. (2007). Metal based drugs: from serendipity to design. Dalton T 43:4903–17.
- Gregoriadis G, Davis C. (1979). Stability of liposomes in vivo and in vitro is promoted by their cholesterol content and the presence of blood cells. Biochem Biophys Res Commun 89:1287–93.
- Guo S, Wang Y, Miao L, et al. (2013). Lipid-coated cisplatin nanoparticles induce neighboring effect and exhibit enhanced anticancer efficacy. ACS Nano 7:9896–904.
- Jehn CF, Boulikas T, Kourvetaris A, et al. (2007). Pharmacokinetics of liposomal cisplatin (lipoplatin) in combination with 5-FU in patients with advanced head and neck cancer: first results of a phase III study. Anticancer Res 27:471–5.
- Johnstone TC, Wilson JJ, Lippard SJ. (2013). Monofunctional and higher-valent platinum anticancer agents. Inorg Chem 52:12234–49.
- Kasparkova J, Vojtiskova M, Natile G, et al. (2008). Unique properties of dna interstrand cross-links of antitumor oxaliplatin and the effect of chirality of the carrier ligand. Chemistry 14:1330–41.
- Kim ES, Lu C, Khuri FR, et al. (2001). A phase II study of STEALTH cisplatin (SPI-77) in patients with advanced non-small cell lung cancer. Lung Cancer 34:427–32.
- Knox RJ, Friedlos F, Lydall DA, et al. (1986). Mechanism of cytotoxicity of anticancer platinum drugs: evidence that cis-diamminedichloroplatinum(II) and cis-diammine-(1,1-cyclobutanedicarboxylato)platinum(II) differ only in the kinetics of their interaction with DNA. Cancer Res 46:1972–9.
- Li Q, Tian Y, Sun J, et al. (2014). The effect of lipocisplatin on cisplatin efficacy and nephrotoxicity in malignant breast cancer treatment. Biomaterials 35:6462–72.
- Maeda H. (2001). The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul 41:189–207.
- Mallik AK, Sawada T, Takafuji M, et al. (2010). Novel approach for the separation of shape-constrained isomers with alternating copolymer-grafted silica in reversed-phase liquid chromatography. Anal Chem 82:3320–8.
- Mizutani Y, Sato N, Kawauchi A, et al. (2002). Cisplatin induced in vivo differentiation of human embryonal carcinoma. BJU Int 89:454–8.
- Nagai N, Ogata H. (1997). Quantitative relationship between pharmacokinetics of unchanged cisplatin and nephrotoxicity in rats: importance of area under the concentration-time curve (AUC) as the major toxicodynamic determinant in vivo. Cancer Chemoth Pharm 40:11–8.
- Nukolova NV, Oberoi HS, Cohen SM, et al. (2011). Folate-decorated nanogels for targeted therapy of ovarian cancer. Biomaterials 32:5417–26.
- Oberoi HS, Nukolova NV, Kabanov AV, et al. (2013). Nanocarriers for delivery of platinum anticancer drugs. Adv Drug Deliv Rev 65:1667–85.
- Paraskar AS, Soni S, Chin KT, et al. (2010). Harnessing structure-activity relationship to engineer a cisplatin nanoparticle for enhanced antitumor efficacy. Proc Natl Acad Sci 107:12435–40.
- Rosenberg B, Vancamp L. (1969). Platinum compounds: a new class of potent antitumour agents. Nature 222:385–6.
- Sengupta P, Basu S, Shicani S, et al. (2012). Cholesterol-tethered platinum II-based supramolecular nanoparticle increases antitumor efficacy and reduces nephrotoxicity. Proc Natl Acad Sci USA 109:11294–9.
- Shaili E. (2014). Platinum anticancer drugs and photochemotherapeutic agents: recent advances and future developments. Sci Progress 97:20–40.
- Uchino H, Matsumura Y, Negishi T, et al. (2005). Cisplatin-incorporating polymeric micelles (NC-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br J Cancer 93:678–87.
- Vail DM, Kurzman ID, Glawe PC, et al. (2002). Stealth liposome-encapsulated cisplatin (SPI-77) versus carboplatin as adjuvant therapy for spontaneously arising osteosarcoma (OSA) in the dog: a randomized multicenter clinical trial. Cancer Chemoth Pharm 50:131–6.
- Wang Y, Zhou J, Qiu L, et al. (2014). Cisplatin-alginate conjugate liposomes for targeted delivery to EGFR-positive ovarian cancer cells. Biomaterials 35:4297–309.
- Wheate NJ, Walker S, Craig GE, et al. (2010). The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans 39:8113–27.
- Xiong Y, Jiang W, Shen Y, et al. (2012). A poly (γ, l-glutamic acid)-citric acid based nanoconjugate for cisplatin delivery. Biomaterials 33:7182–93.