Abstract
Tuberculosis (TB) remains one of the oldest and deadliest diseases in the current scenario. The intracellular organism Mycobacterium tuberculosis, which mainly resides in mononuclear phagocytes, is responsible for tuberculosis in humans. A few therapies are available for the treatment of tuberculosis but they have many hurdles. To overcome these hurdles, a combination of chemotherapeutic agent-loaded vesicular systems have been prepared to overcome tuberculosis. To investigate the role of novel drug delivery systems for the treatment of pulmonary tuberculosis, ligand appended liposomals have been developed. In the present study, drug-loaded, ligand-appended liposomes and their DPI (Dry Powder Inhaler) forms have been prepared and characterized using various in vitro and in vivo parameters. The prepared ligand-appended liposomal formulation showed good entrapment efficiency, prolonged drug release, improved recovery of drugs from the target site, and proved to be more suitable for use as DPI, justifying their potential for improved drug delivery. Thus we tried our best by our project to reduce the national burden of tuberculosis, which is still a global health challenge.
Introduction
The intracellular organism Mycobacterium tuberculosis, which mainly resides in mononuclear phagocytes, is responsible for tuberculosis in humans (Alcais et al. Citation2005). Therefore there is a need to strengthen tuberculosis treatment, its control, and to develop a new approach for tuberculosis (TB) treatment (Gandhi et al. Citation2006). The problem of drug resistance is dominant in cases of tuberculosis, and problems like inefficient drug delivery and off-target biodistribution have made tuberculosis treatment a challenge (Smith Citation2003, Brosch et al. Citation2002). Our macrophage and RES systems recognize formulation as foreign and try to degrade it. Target drug distribution is another aspect that must be achieved at any cost for successful therapy. Different approaches have been attempted to overcome these problems by providing “selective” delivery to the affected area. It is therefore desirable that, by using available optional strategies, differentiated concentration should be produced selectively higher in cellular tropics of infection compared to the blood plasma pool (Vyas and Khatri Citation2007). These can be achieved using aerosolized liposomal systems delivered directly to the macrophages, resulting in an increase in ability for the target site and to fight against disease (Churchyeard et al. Citation2007). To overcome these hurdles, a combination of ligand-appended, drug-loaded vesicular systems have been prepared to overcome the tuberculosis problem. In our present investigation, our aim is to address the problems of intrapulmonary delivery of ligand-appended chemotherapeutic agents in the form of DPI to overcome drug-resistant tuberculosis and to improve the efficacy of formulation. Ligand-appended liposomes were found to be one of the most extensively investigated systems for controlled drug delivery to the lungs, since they can be prepared from the phospholipids endogenous to the lungs as surfactants (Hopewell Citation1994, Kamholz Citation1996). The principal liposome components, phospholipids and cholesterol, are processed by normal metabolic and elimination routes and have low toxicities, the presence of cholesterol and phospholipids with saturated hydrocarbon chains, and increase the drug residence time within the lungs as they decrease liposomal membrane permeability to the encapsulated drug and protect liposomes from in-vivo destabilization (Mehta et al. Citation1996, Noss et al. Citation2001).
Egg phosphatidylcholine- and cholesterol-based liposomes were modified by coating them with alveolar macrophage-specific ligands like mannan, and O-steroyl amylopectin [O-SAP]) (Via et al. Citation1997, Ferrari et al. Citation1999). Mannan have been used as ligands for drug delivery to the receptor site. Rifampicin (RIF), isoniazid (INH), and pyrazinamide (PYZ) were the drugs chosen for the preparations of neutral and ligand-appended liposomes and their DPI forms (Conte et al. Citation2000). Rifampicin was lipophilic in nature while isoniazid and pyrazinamide were hydrophilic in nature.
Thus, the aim of the present study was the development and characterization (in-vitro and in-vivo) of ligand- appended liposomes for multiple drug therapy for pulmonary tuberculosis.
Material and methods
Instrumentation
Spectral and absorbance measurements were made on a UV spectrophotometer (Shimadzu, Japan, UV 1700 Pharma Spec). A digital balance (Mettler Toledo AG, Laboratory & Weighing Technologies, Switzerland, AB265- S/ FACT) was used for weighing the samples. An incubator shaker (Daihan Labtech. Co. Ltd., Korea, LSB- 1005RE) was used for the drug release study. A particle size analyzer (Beckman Coulter Pvt. Ltd., Delsa NanoC) was used for particle size and zeta potential measurement.
Chemicals
Rifampicin (RIF), Isoniazid (INH), and Pyrazinamide (PYZ) were taken as gift samples from Lupin Pharmaceutical Pvt. Ltd., Aurangabad, India. Phosphatidylcholine (PC), cholesterol (CHOL), dialysis tubing, Sephadex G-50, and mannan were purchased from Sigma (USA). Chloroform, lactose, and heparin were purchased from CDH Laboratory Reagent, New Delhi, India. Dimethyl sulfoxide (DMSO), disodium hydrogen phosphate, D-mannitol, methanol and potassium dihydrogen phosphate were purchased from Rankem Laboratory Reagent, New Delhi, India. Triton X-100 was purchased from Himedia Laboratories Pvt. Ltd., Mumbai, India. All other chemicals and solvents used were of analytical grade.
Preparation of neutral and ligand-appended liposomes
Neutral liposomes were prepared by the thin film hydration method. Constitutive lipids (PC:Chol; 7:3 molar ratio) were dissolved in minimum volume of chloroform. A known amount of drug dissolved in organic solvent was added to the lipid solution. Organic solvent was slowly removed under reduced pressure, using a rotary flash evaporator at 150 rpm for 45 min. in a thermostatically controlled water bath at 37°C under vacuum of 600 mm Hg, so that a thin film of dry lipid was deposited on the inner wall of the flask. After film formation, the RBF was hydrated with 5 mL of PBS (pH7.4) for 3 hrs (Mansour et al. Citation2009, Jain Citation2008, Pandy et al. Citation2009). Thus, neutral liposomes of rifampicin (RIF), isoniazid (INH), and pyrazinamide (PYZ) were prepared using this procedure. Mannan was used as a ligand attached with the liposomes. The incubation method was used for the attachment of mannan to the liposomes. Coating solutions were prepared by dissolving mannan (1:1) w/w in hot water. Coating was done by depositing these coating solutions on the surface of liposomes by mixing the preformed liposomes with mannan solution and stirring this mixture overnight at room temperature. Free mannan was removed by passing through a sephadex G-50 column. The methods of preparation of mannan-coated liposomes were the same as those for neutral liposomes.
Characterization parameters (in vitro and in vivo)
Size and shape
The mean vesicle diameter of the prepared plain and polymer-coated vesicles was measured using an optical microscope (Leitz-Biomed, Germany) at 100×magnification with the help of a stage micrometer. The shape of the vesicles was also studied using the optical microscope.
Particle size and zeta potential
The mean particle size and zeta potential of the prepared liposomes were measured using a particle size analyzer. The average particle size and zeta potentials were measured after performing the experiment in triplicate. The particle size analyzer (Delsa nano series) is a new generation of instruments that use photon correlation spectroscopy (PCS), which determines particle size by measuring the rate of fluctuations in laser light intensity scattered and zeta potential works on the principle of electrophoretic light scattering (ELS), which determines electrophoretic movement of charged particles under an applied electric field from the Doppler shift of scattered light for zeta potential determination.
Entrapment efficiency
The free un-entrapped drug was removed by passing the dispersion through a Sephadex G-50 column. The vesicles were disrupted using 1.0 ml of 0.1% (v/v) Triton X-100 and the liberated drug was estimated spectrophotometrically using a UV-VIS spectrophotometer.
Entrapment efficiency (%) = [(A2B)/A]*100
Where A is concentration of total drug and B is concentration of free drug.
In-vitro release profile
In-vitro release rate was determined by using dialysis bags (Sigma, USA). One ml of the prepared plain/ligand-appended vesicle was taken and both ends of “the dialysis bag” were tied. The bag was placed in a beaker containing 250 ml PBS, pH 7.4. The beaker was placed over a magnetic stirrer (York, India) and the contents were stirred at 37°C ± 2°C at a constant speed. Aliquots of samples were withdrawn at regular intervals and thereafter at 24 h. After each sampling time, the volume (5 ml) of receptor compartment was replaced with an equal volume of fresh PBS pH 7.4. The drug content in the withdrawn samples was estimated spectrophotometrically; cumulative percent of drug released was calculated and plotted against time (t).
Evaluation of release kinetics
Various models (zero-order, first-order, higuchi model and Korsemeyer-Peppas) were applied in order to evaluate the release data. Regression coefficient (R2), elimination rate constant, and diffusion exponent (n) were calculated and proximity to 1 for (R2) and 0.45 for (n) was the reference.
Preparation of dry powder inhaler (DPI)
The liposomal dispersions containing sucrose as a cryoprotectant were frozen at –40°C and lyophilized for 48 hours and the porous cakes thus formed were obtained. Similarly, the lyophilized liposomal powder was mixed with a lactose carrier in mass ratio of 1:5 (Sally-Ann Citation2005, Yvas and Sakthivel Citation1994, Papahadjopoulos et al. Citation1973). The bottles were stored in desiccators at refrigeration temperature (2°C–8°C) until further use.
Characterization parameters
Angle of repose
It was determined by the fixed funnel method. The pile of powder was carefully built up by dropping the powder material through a funnel tip from height of 2 cm. The angle of repose was calculated by inverting tangentially the ratio of height and radius of the formed pile.
Moisture content
The moisture content of the dry powders was performed by the Karl Fisher volumetric titration method.
Morphology
The lyophilized powder was coated with gold and then kept in the sampling unit as a thin film and the photographs were taken at different magnifications using scanning electron microscopy (SEM).
In-vivo studies
The developed lyophilized liposomal powders were studied for tissue distribution. In-vivo studies were performed on adult albino rats by administering various lyophilized products through a suitable device.
Organ homogenate studies
Animals were exposed to various formulations and were sacrificed after 1, 2, 8, and 24 h. Visceral organs (liver, spleen, lung, and kidney) of the dissected rats were removed, washed to remove any adhered debris, and dried using a tissue paper. The isolated organs were weighed separately and homogenized in a phosphate buffer using a tissue homogenizer. The homogenized tissues (20%) were deproteinized with 100 µl of acetonitrile, kept in the dark for 30 min, and filtered. To the filtrate, 100µl of extraction fluid methanol was added with vigorous shaking and then centrifuged at 5000 rpm for 30 minutes. The supernatant was separated and drug content was measured using a UV-VIS spectrophotometer. The supernatants from successive extracts of an organ from each rat were pooled and the drug content was determined. The amount of drug in each organ was calculated as percent drug recovered from the respective organ at different time intervals.
Results
Targeting ligand-anchored liposomes bearing rifampicin, isoniazid, and pyrazinamide to the lungs and, specifically, to the pulmonary alveolar macrophages by DPI, is a promising and feasible strategy for treating mycobacterial infections. The selected ligand for the present study was mannan, which has specific affinity for macrophage mannan receptors. Considering its exquisite specificity and the abundance of its expression, the macrophage mannan receptor and ligand system were chosen for targeted rifampicin, isoniazid, and pyrazinamide delivery to macrophages.
Initially, neutral liposomes were prepared and experiments were conducted to determine the optimized ratio of lipid:cholesterol, which was found to be 7:3 using different concentrations of PC:CHOL (Phosphatidylcholine: Cholesterol) by keeping the drug constant; i.e., for rifampicin drug was 5mg, for isoniazid drug was 2mg, and for isoniazid drug was also 2mg. The optimized results are given in .
Table I. Optimized ratio along with characterization parameters.
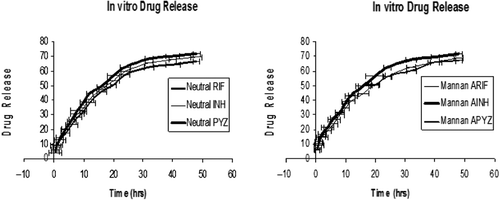
After this, experiments were conducted to determine the amount of the ligand (mannan) which was anchored to the prepared optimized liposomes. For this different concentrations of mannan, i.e. 2 mg, 4 mg, and 6 mg, were appended to the optimized liposomes and 4 mg concentration was selected because it gave the best results, as shown in . It was found that, on addition of 2 mg or 6 mg concentration of mannan, parameters such as zeta potential (ZP), Polydispersity Index (PDI), particle size (PS), and entrapment efficacy (EE) were found to be either high or low. On the other hand, on addition of 6mg mannan concentration, all parameters gave the best result; e.g., less particle size, good entrapment efficacy, less PDI, and good zeta potentials were obtained. Parameters for the selection of optimized mannan concentration are given in along with optimized results. Entrapment of rifampicin in the liposomes could be attributed to the lipophilic nature of the drug represented by lipid:aqueous phase ratio and acyl chain length of phospholipid. Due to the lipophilic nature of the rifampicin, it probably gets intercalated preferentially into the multilamellar vesicle lipid domains. Entrapment of isoniazid and pyrazinamide in the liposomes could be attributed to the hydrophilic nature of the drug represented by the lipid:aqueous phase ratio and acyl chain length of phospholipid. An increase in chain length of fatty acid and inclusion of cholesterol results in an increase in the encapsulation efficiency. The drug molecules may also influence liposome formation by an ability to bind water molecules and form liquid crystal structures in aqueous solutions. Also, all optimized formulations of neutral and ligand-appended liposomes were also characterized for in-vitro drug release study and their in-vitro drug release pattern showed sustained release over 48 hr. It was found that neutral liposomes containing rifampicin, isoniazid, and pyrazinamide showed 65.3 ± 1.67%, 63.2 ± 2.98%, and 57.9 ± 3.78% drug release, and followed the Korsemeyer-Peppas Model, respectively. Similarly, mannan-appended liposomes containing rifampicin, isoniazid, and pyrazinamide showed 67.4 ± 2.43, 65.8 ± 2.56 and 58.1 ± 1.65% drug release, and also followed the Korsemeyer-Peppas Model, respectively, as shown in . These results showed that our preparations follow a controlled release pattern of the drug.
Table II. Optimized mannan concentrations along with characterization parameters.
Figure 1. In-vitro drug release of neutral and mannan-coated liposomes of drugs where manna ARIF, AINH, APYZ represent mannan-appended liposomes of RIF, INH, and PYZ.
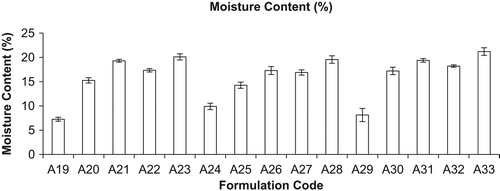
The optimized liposomal formulation of conventional and mannan-coated liposomes was lyophilized using various cryoprotectants for the percent drug retained (PDR). It was found that liposomes were best preserved in their structure with PDR using sucrose as a cryoprotectant in mass ratio of lipid: sucrose at 1:4 (PDR of RIF, 59.97 ± 0.003%, PDR of INH, 59.73 ± 0.033, and PDR of PYZ, 58.26 ± 0.013), as given in . During the freeze-drying process of liposomes, they constrict and get coated on the optimum surface of crystallized sugar. Hydration of polar head groups with the hydroxyl group of sucrose leads to stabilization of liposomes. If the sucrose concentration is less than optimum, the crystallized sugar does not provide adequate surface for the adherence of the constricted bilayer, leading to drug leakage. Hence, the bulk concentration of sugar required as a cryoprotectant depends on the type of sugar selected and saturation of the polar head groups of the bilayer by drug or other formulation components. Lyophilized liposomes keeping the liposome: lactose weight ratio of 1:1 were evaluated, as high-energy adhesion sites (HA) of lactose may bind strongly to the carrier and low-energy adhesion sites (LA) may allow the formation of more reversible bonds with liposomal drugs. This action results in efficient detachment of the liposomal drug from the carrier, as observed with plain DPI formulations. Liposomal drug powder adheres to carrier particles, as seen in SEM photomicrographs of mannan-coated RIF, INH, and PYZ-LDPI (lyophilized DPI) formulations. Evaluation and control of flow and dispersion (de-aggregation) characteristics of the formulation are of critical importance in the development of DPI products. Inter-particle forces that influence flow and dispersion properties are particularly dominant in micronized or microcrystalline powders required for inhalation therapy (< 5 μm). It has been demonstrated that powder adhesion, mediated in part by Van der Waal forces, is directly related to particles < 10 μm. Flow and dispersion properties such as angle of repose and moisture content were characterized as shown in and . The flowability and floodability expressed by angle of repose were found to be 28.15 ± 2.40 (A19), 25.25 ± 1.26 (A20), 19.30 ± 1.40 (A21), 23.42 ± 0.76 (A22), 18.79 ± 1.03% (A23), 29.90 ± 1.57% (A24), 24.25 ± 0.55% (A25), 20.30 ± 1.34% (A26), 24.89 ± 1.2% (A27), 20.50 ± 1.8% (A28), 30.12 ± 1.6% (A29), 25.21 ± 0.76% (A30) and 21.29 ± 1.78% (A31), 23.45 ± 1.45% (A32) and 19.34 ± 1.96% (A33) and moisture content was found to be 07.25 ± 0.41% (A19), 15.25 ± 0.54% (A20), 19.30 ± 0.30% (A21), 17.34 ± 0.34% (A22), 20.10 ± 0.64% (A23), 09.90 ± 0.67% (A24), 14.25 ± 0.65% (A25), 17.30 ± 0.84% (A26), 16.89 ± 0.54% (A27), 19.56 ± 0.78% (A28), 08.12 ± 1.35% (A29), 17.12 ± 0.76% (A30), 19.39 ± 0.38% (A31), 18.21 ± 0.25% (A32), and 21.20 ± 0.79% (A33), respectively. Moisture content determination is also important for drug stability upon storage and deaggregation upon inhalation. Incorporation of CHOL is known to cause strong reduction in the permeability of the liposome system and thus reduce leakage of drug from the liposomes. However, under the present anhydrous state of storage, the incorporation of CHOL reduces the permeability of the membrane, because in the anhydrous state there is not any possibility of drug diffusion; therefore, drug retention cannot be increased by reducing the permeability alone. CHOL have some association with EPC at the molecular level through weak bonding, thereby reducing the level of stress vector introduced during lyophilization.
Table III. Percent (%) drug retention (PDR) study.
Figure 2. Formulation codes and angle of repose (n = 3, mean ± S.D.).
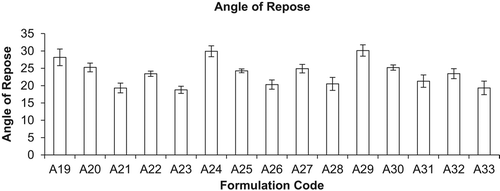
Figure 3. Moisture content (%) of different formulations (mean ± S.D., n = 3). Here A19, A24, and A29 represent inhalation powder of drugs; A20, A25, and A30 represent neutral liposomes of drugs for inhalation; A21, A26, and A31 represent DPI of neutral liposomes of drugs; A22, A27, and A32 represent mannan-coated liposomes of drugs; and A23, A28, and A33 represent DPI of mannan-coated liposomes of drugs, i.e. RIF, INH, and PYZ, respectively.
In-vitro drug release of various lyophilized formulations was monitored for 48 hrs and it was observed that all the formulations showed a controlled release pattern. Different formulations were characterized for in-vitro drug release including DPI of neutral lipsomes of rifampicin, isoniazid, and pyrazinamide, and DPI of mannan-coated liposomes of rifampicin, isoniazid, and pyrazinamide. It was observed that DPI of mannan-coated liposomes showed significant controlled release of drugs as compared to the DPI of neutral liposomes and following the Korsemeyer-Peppas Model.
Further, in- vivo studies were performed on Wistar albino rats through the pulmonary route. An organ tissue distribution study of the developed lyophilized liposomal formulations was compared with the control (fine drug powder). Firstly, DPI of neutral liposomes of rifampicin, isoniazid, and pyrazinamide were given in combinations following a single pulmonary administration, i.e. a combination of RIF + INH + PYZ (2 + 1 + 1 mg); similarly DPI of mannan-coated liposomes, i.e. a combination of RIF + INH + PYZ (2 + 1 + 1 mg), was given to rats. After this the drug was detected in the lungs, liver, spleen and kidney from 1 hr onwards up to 24 hrs. At each time point the drug concentrations were at or above the minimum inhibitory concentration (0.2µg/ml). Groups of Wistar albino rats were canulated, i.e. canula (having the powder) was inserted into the lungs. It is further revealed that, in this study, the drug distribution profile was observed in healthy rats. All the developed liposomal formulations showed greater accumulation in the lungs when compared with the control. In the case of free drug inhalation, i.e. powder of rifampicin, isoniazid, and pyrazinamide, only 34.12%, 37.34%, and 36.76% of the administered dose was found in the lungs at 1 hr post-administration and was not detectable in the lungs after 24 hr.
The lung uptake of the DPI of mannan-coated liposomes was found to be higher compared to the DPI of neutral liposomes. Neutral DPI showed an initial lung accumulation (48.66% after 1 hr) and after 24 hr drug concentration in the lung was found to be 06.46%, but in the case of DPI of mannan-coated liposomes showed higher drug accumulation of 51.36 after 1 hr, and after 24 hr the drug found in the lung was 09.63%. shows the tissue distribution of drugs after administration of different liposomal formulations. represents the SEM images of DPI of RIF, INH, and PYZ.
Table IV. Tissue distribution of drugs after administration of different liposomal formulations.
Figure 4. SEM images of DPI of mannan-coated liposomes of RIF (A23), INH (A28), and PYZ (A33).

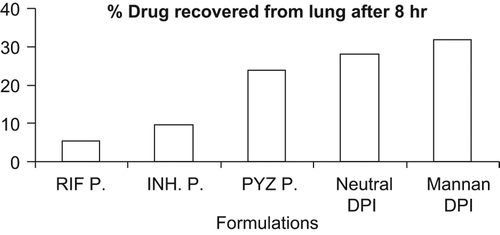
They readily distributed to systemic circulation, from where they were rapidly taken up by the liver and spleen and cleared (digested) by the tissue macrophages there. This may be due to selective targeting of mannan-coated liposomes to the target site, i.e. alveolar macrophages. The comparative percent (%) drug recovered from the lungs after 8 hrs from all the formulations is given in . The observed values suggest that the DPI of mannan-coated liposomes were not only effective in rapid attainment of high-drug concentrations in alveolar macrophages (lungs), but could also maintain the concentration over a prolonged period of time when compared to the free drug. Mannan-coated DPI delivered significantly higher amounts of an encapsulated drug to the cells of the reticuloendothelial system, residing mainly in the liver and spleen, than any of the sterically stabilized liposomes. The liposomes could not avoid the first barrier of the reticuloendothelial system localized in the liver; hence they are not taken up by the secondary barrier macrophages localized in the spleen.
Figure 5. Comparative percent drug recovered from lungs after 8-hr administration of various formulations. Here P. represents powder of RIF, INH, and PYZ; neutral DPI represents DPI of neutral liposomes; mannan DPI represents DPI of mannan-appended liposomes.
Discussion
Pulmonary tuberculosis is the most common, with the involvement of lung macrophages containing a large number of tubercle bacilli. Effective chemotherapy for pulmonary tuberculosis can be attained by targeting drugs to lung tissue by tagging specific markers or homing devices onto the surface of liposomes. Liposomes, as well as delivering drugs to the infected site, could also act as drug reservoirs to provide a slow and sustained release of the drug. Ligand-anchored liposomal DPI are not only effective in the rapid attainment of a high drug concentration in the lung (population of AMs), but also in maintaining this over a prolonged period of time. Rifampicin, isoniazid, and pyrazinamide were incorporated in liposomes because the compatibility with the manufacturing method and the perceived benefit of extending it are useful as an anti-tubercular agent. The preformulations study of the drugs showed that rifampicin was found to be highly lipophilic in nature, while isoniazid and pyrazinamide were found to be hydrophilic in nature. The results obtained from the drug release study showed that the preparations, i.e. conventional and mannan-coated liposomes and their DPI forms, follow a controlled and sustained release pattern of the drugs.
In the present study, we observed the development and evaluation (in-vitro and in-vivo) of the vesicles encapsulating RIF, INH, and PYZ combinations. Different lipid concentrations were used during the formulation to find the best results in terms of particle size, entrapment efficiency, zeta potential, polydispersivity index, and in-vitro drug release. Formulations with ratio 7:3 and having 4mg mannan concentration were found to be optimized, and they formed spherical liposomes. After this dry powder inhaler (DPI) of neutral and ligand (mannan)-appended liposomes were prepared and their characterization was done using parameters like angle of repose, moisture content, and in-vitro drug release. It was found that liposomes were best preserved in their structure using sucrose as a cryoprotectant in mass ratio of lipid: sucrose at 1:4. Lactose was revealed to be a good carrier in the DPI. Preparations were successfully prepared and stabilized by lyophilization in DPI formulations. The in-vivo experiments on adult albino rats led us to conclude that optimized mannan-appended DPIs could be a suitable system for the inhalation of drugs. However, more detailed studies using infected animal models are needed to better explain the macrophage uptake profile of formulations and the targeting activity of liposomal formulations. The results of the in-vivo study showed that percentage dose recovered from DPI of mannan-coated liposomes was significantly higher when compared to the DPI of neutral liposomes, which could be due to maintenance of the concentration of the drug within the therapeutic effective range for a longer period of time from the encapsulated liposomal systems. The liposomal delivery systems may reduce the toxicity of the drugs. DPI of rifampicin, isoniazid, and pyrazinamide loaded liposomes were directly administered to the lungs in combination and were compared with DPI of mannan-coated liposomes, potentially circumventing the toxicity resulting from large oral doses. It is not suggested that it is a clinically relevant efficacy model, but it is a necessary screening tool to evaluate the influence of different mannan concentrations and their characteristics. The residence time of the drug was found to be increased in the lungs in the case of the mannan-coated DPI and their internalization also increased, whereas the free drug was undetectable after 8 hrs after administration through the pulmonary route. Hence, developed liposomal formulation as DPI offers exciting possibilities of liposomal delivery in the anhydrous state. The ability of liposomes to encapsulate a drug within multilamellar vesicles and store the vesicles conveniently in a dehydrated form using a cryoprotectant is advantageous over conventional drug delivery procedures. These values were significantly higher when compared to the administration of the plain drug, which could be due to the maintenance of concentration of drug within the therapeutic effective range for longer periods of time from the encapsulated liposomal systems. The liposomal delivery systems may reduce the toxicity of the drugs. The findings of this investigation demonstrate the possibility of the delivery of liposomally entrapped drugs to the lungs. Hence developed liposomal formulations as DPI offer exciting possibilities of drug delivery in the anhydrous state. Thus, liposomal preparations offer several potential advantages, such as higher patient compliance, direct targeting to the alveolar macrophages, and reduction in systemic side-effects.
Conclusion
Results of this study suggest that encapsulation of anti-tubercular drugs in the liposomes’ modification of the liposomal surface by appending mannan over the liposomes; i.e., using macrophage-specific ligands and deposition to the respiratory tract via DPI will certainly improve the chemotherapy against pulmonary tuberculosis. The strategy on the basis of in-vivo performance appears to be promising. However, it is realized that the work should further be elaborated to study pharmacodynamics of the system(s), viz. macrophage activation profile and their combined role in eradication of M. tuberculosis infection. From this study, we can conclude that a versatile carrier system, i.e. liposome, was found to be an effective tool for the delivery of RIF, INH, and PYZ in combination, and maximum internalization has been achieved into the lungs using mannan as a ligand.
Acknowledgements
The authors are thankful to Lupin Pharmaceutical Pvt. Ltd., Aurangabad, India for gift samples of rifampicin, isoniazid, and pyrazinamide. The help and facilities provided by the Head, Department of Pharmaceutics, ISF College of Pharmacy, Moga Punjab, India, are duly acknowledged.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Alcais A, Fieschi C, Abel L, Casanova JL. 2005. Tuberculosis in children and adults: two distinct genetic diseases. J Exp Med. 202: 1617–21.
- Brosch R, Gordon SV, Marmiesse M, Brodin PC, Buchrieser K, Eiglmeier T, Garnier C, Gutierrez G, Hewinson K, Kremer LM, Parsons AS, Pym S, Samper D, Soolingen V, Cole ST. 2002. A new evolutionary scenario for the Mycobacterium tuberculosis complex.Proc Natl Acad Sci. 99:3684–3689.
- Churchyard GJ, Wallis R, Levin J, Kaplan G, Onyebujoh P, Vahedi M. 2007. Report of the expert consultation on immunotherapeutic interventions for tuberculosis. WHO. Geneva, 1–56.
- Conte JE, Golden JA, McQuitty M, Kipps J, Lin ET, Zurlinden E. 2000. Single dose intrapulmonary pharmacokinetics of rifapentine in normal subjects. Antimicrob Agents Chemother. 44:985–990.
- Ferrari G, Langen H, Naito M, Pieters J. 1999. A coat protein on phagosomes involved in the intracellular survival of mycobacteria. Cell 97:435–447.
- Gandhi NR, Moll A, Sturm AW, Pawinski R, Govender T, Lalloo U. 2006. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet. 368:1575–80.
- Hopewell PC. 1994. Overview of clinical tuberculosis. In Bloom BR. Ed. Tuberculosis: Pathogenesis, Protection, and Control; American Society for Microbiology: Washington, D.C., 25–46.
- Jain KK. 2008. Drug delivery systems–An overview. Methods. Mol Biol. 437:1–50.
- Kamholz SL. 1996. Pleural tuberculosis. In Rom WN and Garay S Eds, Tuberculosis; Little, Brown and Co.:Boston, 483–491.
- Mansour HM, Ree YK, Wu X. 2009. Nanomedicine in pulmonary delivery. Int J Nanomed. 4:299–319.
- Mehta PK, King CH, White EH, Murtagh JJ and Quinn FD. 1996. Comparison of in vitro models for the study of Mycobacterium tuberculosis invasion and intracellular replication. Infect. Immun. 64:2673–2679.
- Noss EH, Pai RK, Sellati TJ, Radolf JD, Belisle J, Golenbock DT, Boom WH, Harding CV. 2001. Toll-like receptor 2-dependent inhibition of macrophage class II MHC expression and antigen processing by 19-kDa lipoprotein of Mycobacterium tuberculosis. J Immunol. 167:910–918.
- Pandey S, Shukla P, Bhatt S, Choudhary V, Viral D, Subhash V. 2009. Microfine dry powder: Pulmonary drug delivery system. J Pharm Res. 2:1159–1162.
- Papahadjopoulos D, Cowden M, Kimelberg H. 1973. Role of cholesterol in membrane; Effect of phospholipid-protein interactions, membrane permeability and enzymatic activity. Biochem Biophys Acta. 330:9–26.
- Sally-Ann C. 2005. Carrier-based strategies for targeting protein and peptide drugs to the lungs. The AAPS Journal. 7: E20–E40.
- Smith I. 2003. Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clinical Microbiology Reviews 16:463–496.
- Via LE, Deretic D, Ulmer RJ, Hibler NS, Huber LA, Deretic V. 1997. Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J Biol Chem. 272:13326–13331.
- Vyas SP, Khatri K. 2007. Liposome-based drug delivery to alveolar macrophages. Expert Opin. Drug Deliv. 4:95–99.
- Vyas SP, Sakthivel T. 1994. Pressurized pack-based liposomes for pulmonary targetting of isoprenaline: Development and characterization. J Microencap. 11:373–380.