Abstract
Background: Catalase catalyzes the reduction of H2O2 to water and it can also remove organic hydroperoxides. Nervous system in body is especially sensitive to free radical damage due to rich content of easily oxidizible fatty acids and relatively low content of antioxidants including catalase. Recent studies indicate that reactive oxygen species actually target active channel function, in particular TRP channels. I review the effects of catalase on Ca2+ signaling and on TRP channel activation in neuroglial cells such as microglia and substantia nigra.
Materials: Review of the relevant literature and results from recent our basic studies, as well as critical analyses of published systematic reviews were obtained from the pubmed and the Science Citation Index.
Results: It was observed that oxidative stress-induced activations of TRPM2, TRPC3, TRPC5 and TRPV1 cation channels in neuronal cells are modulated by catalase, suggesting antioxidant-dependent activation/inhibition of the channels. I provide also, a general overview of the most important oxidative stress-associated changes in neuronal mitochondrial Ca2+ homeostasis due to oxidative stress-induced channel neuropathies. Catalase incubation induces protective effects on rat brain mitochondrial function and neuronal survival. A decrease in catalase activity through oxidative stress may have an important role in etiology of Parkinson’s disease and sensory pain.
Conclusion: The TRP channels can be activated by oxidative stress products, opening of nonspecific cation channels would result in Ca2+ influx, and then elevation of cytoplasmic free Ca2+ could stimulate mitochondrial Ca2+ uptake. Catalase modulates oxidative stress-induced Ca2+ influx and some TRP channels activity in neuronal cells.
| Abbreviations | ||
| ADPR: | = | adenosine diphosphatase ribose |
| CNS: | = | central nervous system |
| FFA: | = | flufenamic acid |
| G3+: | = | gadolinium chloride |
| HEK: | = | human embryonic kidney |
| hST: | = | human syncytiotrophoblast NO: nitric oxide |
| PARG: | = | poly (ADP-ribose) glycohydrolase |
| PARP-1: | = | >poly(ADP- ribose) polymerase |
| ROS: | = | reactive oxygen species |
| SNc: | = | substantia nigra pars compacta |
| SNr: | = | substantia nigra pars reticulate |
| TRP: | = | transient receptor potential |
Introduction
Reactive oxygen species (ROS) are mainly composed of superoxide anion, H2O2 and singlet oxygen. The oxygen-derived species can attack DNA, producing a distinctive pattern of DNA alterations (Citation1). There is also evidence that ROS play an important role in the pathogenesis of many diseases, particularly in neurological diseases due to neurons and brain system vulnerability to oxidative stress (Citation2,Citation3). The existences of polyunsaturated fatty acids which are targets of the ROS in the neuron and brain make this organ more sensitive to oxidative damage (Citation2). Growing evidence suggests that physiological conditions, ROS regulate neuronal signaling both in the central and in the peripheral nervous system (Citation3). However, neurological cells are protected by antioxidants against peroxidative damage (Citation1–7). Antioxidant defenses include the enzymatic and non enzymatic defense systems. Enzymatic defense systems include enzymes superoxide dismutase, glutathione peroxidase, and catalase (Citation4,Citation6). Catalase is a common antioxidant enzyme found in nearly all living organisms that are exposed to oxygen. Catalase catalyzes the reduction of H2O2 to water. Catalase can also remove organic hydroperoxides. In addition, catalase uses H2O2 to oxidize toxins including phenols, formic acid, formaldehyde and alcohols. The brain catalase activity is extremely low compared with other tissue and organs such as liver and kidney (Citation7).
There are differences in antioxidant defense systems in neurological cells (Citation5). For instance, microglial cells are equipped with efficient antioxidant defense mechanisms for self-protection against oxidative damage. These cells contain glutathione in high concentrations, and substantial activities of the catalase as well as NADPH-regenerating enzymes. Their good antioxidative potential protects microglial cells against oxidative damage that could impair important functions of these cells in defense and repair of the brain. However, oligodendrocytes and neurons are less well protected against oxidative stress (Citation6).
The effects of ROS in neuronal cells remain largely unknown. ROS have deleterious effects on various ion channels, such as calcium ion (Ca2+) channels (including voltage gated L-type Ca2+ channels, ryanodine receptor Ca2+-release channels), Ca2+ activated K+ channels, and ATP sensitive K+ channels (Citation3,Citation5,Citation6). Recent studies indicate that ROS actually targets active channel function, in particular transient receptor potential (TRP) channels (Citation4,Citation8,Citation9). Since the discovery of the Drosophlia melanogaster transient receptor potential (TRP) gene, it has emerged that mammalian genomes contain 28 homologous genes encoding proteins that are capable of forming a large variety of homo- or hetero-multimeric cationic channels, usually with permeability to Na+ and Ca2+ (Citation10). The TRP channels superfamily includes TRP canonical (TRPC), TRP vanilloid (TRPV), TRP melastatin (TRPM), TRP polycystein (TRPP), TRP mucolipin (ML), and TRP ankyrin (TRPA) subfamilies. At least eight TRP channels (TRPM2, TRPM7, TRPC5, TRPV1, TRPV3 and TRPV4, TRPM6 and TRPM7) have been found to respond to oxidative stress by increased channel activity (). It is postulated that some TRP channels may have a molecular sensor for oxidative stress. For example, in TRPM2, this is a type of Nudix hydrolase (NUDT9-H) that can bind to and hydrolyze adenosine diphosphate (ADP) ribose (ADPR), although not as effectively as other known Nudix ADPR-hydrolases (Citation11). Binding of ADPR to NUDT9-H activates the channel, allowing the passage of cations down their electrochemical gradient (). Because TRPM2 is a plasma membrane cation channel, Ca2+ and Na+ will flow into the cell when TRPM2 opens. ADPR and ROS are the most potent physiological activators of TRPM2, but other less potent activators have been also proposed (Citation8,Citation12). Catalase induces modulator effects on oxidative stress-induced activation of TRP channels ().
Table 1. Inhibitor role of antioxidant enzyme catalase on TRP channels in different cells activated by oxidative stress.
Figure 1. All member of the TRP channels superfamily, which include TRP cononcial (TRPC) subfamily consist of 7, TRP vanilloid (TRPV) subfamily consist of 6, TRP melastatin (TRPM) subfamily consist of 8, TRP polycystein (TRPP) subfamily consist of 3, TRP mucolipin (ML) subfamily consist of 3, and TRP ankyrin (TRPA) subfamily consist of 1.
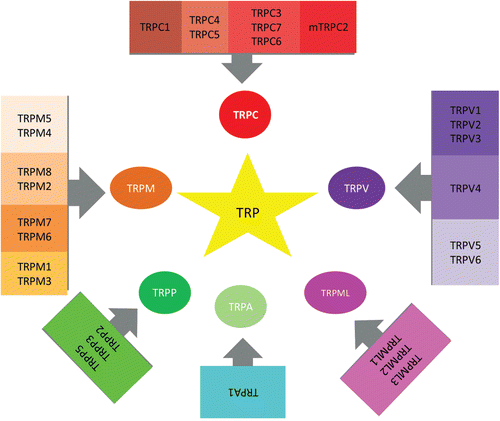
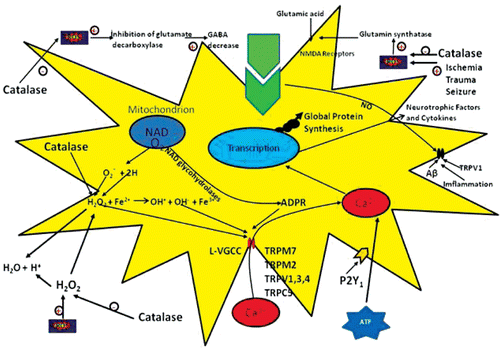
Figure 2. Cells regulate intracellular Ca2+ levels lightly and excessive Ca2+ loads can lead to inappropriate activation of process that are normally operate at low levels, causing metabolic derangements and eventual cell death. For example, excesses elevations in intracellular Ca2+ may activate catalase degradation, induce formation of reactive oxygen species (ROS) or disrupt normal mitochondrial function leading to oxidative stress and bioenergetic failure. ATP acts on astrocytes, which express P2Y1 receptors, to increase the level of intracellular Ca2+. The Ca2+ rise stimulates the release of superoxide radical via activation of NADPH oxidase. In the presence of superoxide dismutase, superoxide radical is dismutased to H2O2 as a diffusible messenger might affect neighboring GABAergic terminals either directly or via dismutation into highly reactive hydroxyl radical (OH). The dismutation of H2O2 into OH is accelerated in the presence of divalent iron via the Fenton reaction. The action of ATP could be blocked either at the astrocyte level by specific P2Y1 receptor antagonists or by catalase, a potent ROS scavenger (Citation21). Excessive Ca2+ load, in particular via N-methyl-D-aspartate (NMDA) receptors, is toxic to neurons in neurodegenerative diseases. NMDA receptor-mediated Ca2+ entry triggers a neurotoxic signal cascade involving the activation of neuronal nitric oxide (NO) synthase, formation of the toxic ROS and NO and activation of the pro-apoptotic protein poly(ADP-ribose) polymerase (PARP-1). Antioxidants regulate Ca2+ influx into cytosol by inhibition of ROS. Sustained depolarization of mitochondrial membranes and enhanced ROS production activates transient receptor potential (TRP) channels such as TRP melastatin 2 (TRPM2), TRP vanilloid (TRPV), TRP cononcial (TRPC) and voltage gated Ca2+ channels (VGCC) and Ca2+ influx increases by the activation of TRP via ROS. The molecular pathway may be a cause of neurological symptoms and represents a fruitful subject for further study.
Oxidative stress and Ca2+ signaling in neuronal cells
Ca2+ions pass the cell membrane through various ion channels such as voltage gated Ca2+ channels, receptor operated Ca2+ channels, store operated Ca2+ channels and TRP channels. Also, Ca2+ extrusion through a plasma membrane Ca2+ pump is decreased in neuronal diseases. Ca2+ is released from the ER stores through both the inositol trisphosphate (InsP3) and ryanodine receptors (Citation13).
Both endoplasmic reticulum and mitochondria are organelles intimately connected with Ca2+ homeostasis, with endoplasmic reticulum being able to participate in neuronal Ca2+ signaling, either as an amplifier of Ca2+ release or as a sink (Citation14,Citation15). Mitochondria are traditionally seen as the low-affinity, high capacity Ca2+ sink, preventing excessive cytosolic Ca2+ load, but it is clear that their participation in Ca2+ homeostasis and in physiological processes dependents on Ca2+ is much wider (Citation14,Citation16).
ROS including superoxide anion, H2O2 and singlet oxygen act as subcellular messengers in such complex processes as mitogenic signal transduction, gene expression, and regulation of cell proliferation when they are generated excessively or when enzymatic and non-enzymatic defense systems are impaired (Citation2,Citation4). While many intra- and extracellular molecules may participate in neuronal injury and cell apoptosis, accumulation of oxidative stress due to excessive generation of ROS appears to be a potential factor for cell damage and death (Citation16). Growing evidence suggests that, in physiological conditions, ROS regulate neuronal signaling in both the central and peripheral nervous system. Although at the periphery, ROS contribute to the inhibitory effects of ATP on quintal acetylcholine release from motor nerve ending, in the central nervous system, ROS produce both enhancement and depression of synaptic transmission (Citation17).
In the nervous system, ROS are produced mainly by microglia (Citation18) and by astrocytes (Citation19). In cultured astrocytes, activation of P2Y receptors by extracellular ATP induces ROS via membrane-bound NADPH oxidase (Citation20). ATP acts on astrocytes, which express P2Y1 receptors, to increase the level of cytosolic Ca2+. The Ca2+ rise stimulates the release of superoxide radical via activation of NADPH oxidase. In the presence of superoxide dismutase, superoxide radical is dismutated to H2O2 as a diffusible messenger might affect neighboring GABAergic terminals either directly or via dismutation into the highly reactive hydroxyl radical. The dismutation of H2O2 into hydroxyl radical is accelerated in the presence of bivalent iron via the Fenton reaction. A single astrocyte may control ~140,000 synapses, providing widespread control of synaptic transmission. The action of ATP can be blocked either at the astrocyte level by specific P2Y1 receptor antagonists or by catalase, a potent ROS scavenger (Citation21). Safiulina et al. (Citation21) reported neither N-acetyl cysteine (100 μM) nor catalase (1200 units/mL) prevented Ca2+ transients induced by ATP in astrocytes.
H2O2 is also generated during β-amyloid aggregation (Citation3), dopamine oxidation (Citation22), and brain ischemia/reperfusion (Citation3). The released H2O2 is readily converted into the highly toxic hydroxyl radical by the Fenton reaction and causes further damage to lipids, protein and nucleic acids. Such oxidative damage can lead to mitochondrial dysfunction, Ca2+ imbalance and apoptosis in neuronal cells (Citation23). Recent studies showed that H2O2 induces cytotoxicity in neuronal cells such as PC12. The damage includes impairment of cell membranes and the nucleus. There is also a decrease in mitochondrial membrane potential and in certain antioxidant enzyme activities such as those catalase and glutathione peroxidase, an increase in ROS level, and depletion of glutathione (Citation24).
Cytosolic Ca2+ has been presented as a key regulator of cell survival but this ion can also induce apoptosis in response to a number of pathological conditions () (Citation3,Citation4). In addition, the mitochondria acts as Ca2+ buffers by sequestering excess Ca2+ from the cytosol (Citation23). Ca2+ mobilizing agonists can effectively produce a rapid, simultaneous and reversible cessation of the movements of both endoplasmic reticulum and mitochondria, which is strictly dependent on a rise in cytosolic free Ca2+ concentration. This inhibition in mitochondrial motility reflects an increased mitochondrial Ca2+ uptake and thus enhances the local Ca2+ buffering capacities of mitochondria, with important consequences for signal transduction (Citation26). Numerous reports indicate that ROS at physiological concentrations in mitochondria of human leukemia 60 cells and human leucocytes act as requisite signaling molecules in processes underlying neuronal functions (Citation4,Citation26,Citation27). The redox state of the mitochondria modifies the activity of several molecules involved in Ca2+ signaling although antioxidants such as melatonin induced protective effects on the Ca2+ signaling (Citation27). Ca2+ overloading of mitochondria can also induce an apoptotic program by stimulating the release of apoptosis promoting factors such as cytochrome c, and by generating ROS due to respiratory chain damage (Citation23,Citation28). The release of Ca2+ from endoplasmic reticulum stores by IP3 receptors has been also implicated in multiple models of apoptosis by being directly responsible for mitochondrial Ca2+ overload (Citation14). Recently, we provided compelling evidence to support the belief that mitochondrial Ca2+ uptake through TRPM2 cation channels evoked by rises in cytosolic free Ca2+ concentration induces mitochondrial membrane depolarization. Our recent results indicated that blockade of both Ca2+ uptake into mitochondria with thiol group antioxidants such as N-acetyl cysteine (Citation25) and glutathione (Citation11) or rises in cytosolic free Ca2+ concentration in dorsal root ganglion neuron through voltage gated and TRPM2 Ca2+ channels were able to decrease oxidative stress mediated by H2O2, which was able to block Ca2+ release from intracellular stores. The toxic properties of oxidative stress with regard to the function of TRPM2 channels in catalase depleted neurons such as those of the dorsal root ganglion, however, still remain unknown.
Changes in mitochondrial integrity, ROS release and Ca2+ handling are also proposed to be involved in the pathogenesis of methylmalonic acidaemia and Huntington’s disease, which exhibit partial mitochondrial respiratory inhibition. Catalase prevents mitochondrial permeability transitions by removing mitochondrial H2O2 (Citation28). Macial et al. (Citation29) investigated the mechanisms by which inhibitors of the respiratory chain complex II and catalase affected rat brain mitochondrial function and neuronal survival. Catalase (1 μM) partially inhibited or prevented the loss of mitochondrial depolarization, suggesting that the inner mitochondrial membrane permeabilization under these conditions is at least partially caused by accumulation of mitochondrial H2O2. Vesce et al. (Citation30) reported that potent cell-permanent superoxide dismutase/catalase mimetic manganese tetrakis (N-ethylpyridinium-2yl) abolished deregulate on-related increase in superoxide radical of single cultured rat cerebellar granule neurons exposed continuously to glutamate and the deregulation of cytoplasmic Ca2+. A combination of catalase (250 IU/mL) with free radical scavenger 4-hydroxy-TEMPO also fails to reduce deregulation.
Transient receptor potential (TRP) Superfamily
The TRP cation channel superfamily is compromised of a diverse range of voltage-dependent Ca2+-permeable cation channels. There are 28 mammalian TRP channels, grouped into seven subfamilies. Unlike many other types of ion channel, the activity is not gated by changes in voltage and there is no fast neurotransmitter-dependent gating. Instead, the channels respond relatively slowly to various chemical and physical factors. In the case of some types of TRP channel, there are extensive knowledge of the chemical-sensing capabilities, and roles as sensors of chemical or temperature changes in the environment have been proposed (Citation11).
The TRP channels superfamily includes 7 TRPC subfamily members, 7 TRPV subfamily members, 6, TRP TRPM subfamily members, 8 TRPP types, 3 TRP ML subfamily members and 3 and TRPA subfamily members. This last subfamily is poorly characterized, but is attracting increased interest because of their involvement in several human diseases (Citation31). TRP channels have basic structures similar to voltage-gated potassium channels, with homo- or hetero-tetrameric arrangements around a central ion conducting pore between the 5th and 6th transmembrane segments of the channel pore (Citation32). The 4th transmembrane segment is not positively charged. The N-termini of TRPV and TRPC, but not those of TRPM channels, contain multiple ankyrin binding repeats (Citation33). The C-terminal part of the 6th transmembrane segment in TRPC and TRPM channels includes the ‘TRP domain’, which is a conserved stretch of 25 amino acids starting with the nearly invariant ‘TRP box’ that is missing in TRPV channels (Citation10).
Role of TRPM2 channels in neurological cells
TRPM2 is expressed in diverse cell types and despite convincing evidence for high expression in the mammalian brain, much of this signal was attributed to strong expression in non-neuronal cells. Thus, the existence of functional TRPM2 channels in neurons is controversial at best. Olah et al. (Citation34) reported that functional TRPM2 channels are highly expressed in pyramidal neurons of the hippocampus, including those of CA1 interneurons in hippocampal slicas. These authors also reported that ADPR alone is insufficient to gate TRPM2 in hippocampal neurons. They concluded that concomitant influx of Ca2+ through voltage dependent Ca2+ channels and/or NMDAR is necessary to fully active TRPM2 channels. Xie et al. (Citation35) reported in CA1 pyramidal neurons of the hippocampus that the activation of TRPM2 channels may be sensitized by co-incident NMDA receptor activation, suggesting a potential contribution of TRPM2 to synaptic transmission. Recently, we observed oxidative stress-dependent activation of TRPM2 channels in dorsal root ganglion neurons and the activated channels were modulated by antioxidants such as glutathione (Citation36) and by indirect channel blockers such as aminoethoxydiphenyl borate and flufenamic acid (FFA) (Citation11,Citation37).
Catalase and TRP channels in substantia nigra and Parkinson’s disease
Parkinson’s disease is a neurodegenerative disease that selectively targets dopamine neurons of the substantia nigra pars compacta (SNc) and reticulate (SNr). Mitochondrial dysfunction has been implicated as an important source of oxidative stress (Citation3). Key evidence includes decreased activities of mitochondrial complex I in SNc and other tissues of patients with Parkinson’s disease (Citation22). Partial inhibition of complex I in isolated mitochondria causes increased production of ROS, especially H2O2 (Citation15). These observations have led to the development of a rodent model of Parkinson’s disease based on chronic exposure to the mitochondrial complex I inhibitor, rotenone. Rotenone is a naturally occurring isoflavonoid from the tropical plants Lonchocarpus and Derris (Citation38). Rotenone decreases intracellular ATP levels and increases the production of ROS including superoxide anion, H2O2 and singlet oxygen (Citation39). It also releases glutamate presynaptic terminals, leading to an additional increase in ROS production (Citation40). Mitochondrial derived ROS can also activate TRPM2 channels (Citation32). As TRPM2 channels are permeable to Ca2+ and have previously been implicated in other neurodegenerative disorders (Citation41), activation of these channels is a potentially important mechanism that may contribute to the pathogenesis of DRG and SNc neuron dependent pain and disorders (Citation40).
Bao et al. (Citation42) tested whether inhibition of axonal dopamine release by rotenone involved H2O2 by using the H2O2-metabolizing enzyme, catalase (500 U/mL). In their study, implicating a key role for H2O2, the effects of 50 nM rotenone on single pulse evoked dopamine currents were completely abolished by catalase. However, heat-inactivated catalase did not alter rotenone-induced suppression of dopamine release. In addition, rotenone-induced changes in membrane properties occurred in the absence of a change in tissue ATP and were prevented by catalase, as well as by the TRPM2 channels blocker, FFA, indicating that brain TRP channels like KATP channels, are potential targets for regulation by endogenous H2O2.
It is likely that TRPC3 is also directly activated in response to oxidative stress (Citation43). The oxidant tert.-butylhydroperoxide completely depolarized endothelial cells by the activation of a TRP-related cationic current and expression of a dominant negative N-terminal splice variant of TRPC3 (Citation44). These results suggest that the TRPC protein determines endothelial redox sensitivity and suppression of TRPC, could be an approach to the treatment of oxidative stress-induced vascular dysfunction. In addition, over expression of TRPC3 in HEK293T cells showed an increase in basal membrane conductance upon tert.-butylhydroperoxide treatment, which is mainly due to the influx of Na+ (Citation45). This suggests that TRPC3 and TRPC4 form redox sensitive cation channels during oxidative stress and could participate in Na+ loading and membrane depolarization. Hence, activation of TRPC channels in SNc neurons increases the firing rate of these cells and contributes to tonic depolarization that maintains firing.
To determine whether basal levels of endogenous H2O2 generated in SNr GABAergic neurons during spontaneous activity influence firing rate, Lee et al. (Citation46) depleted endogenous H2O2 using catalase (500 U/mL). Catalase caused a 40% decrease in the spontaneous firing rate of SNr neurons. They concluded that the basal H2O2 level modulates the rate and regularity of spontaneous activity of SNr GABAergic neurons. Fenamates like FFA are anti-inflammatory drugs known to alter ion fluxes through the plasma membrane. They are for instance, potent blockers of cation and anion channels, and FFA at ≥50 μM is now commonly used to block currents through TRP channels and receptor-operated channels (Citation40). Previous experiments have shown that tonic activation of TRP channels maintains basal firing rate, as well as the regularity of firing, in SNr neurons (Citation47). Lee et al. (Citation46) tested whether the decrease in firing rate caused by application of the non-selective TRPC and TRPC3 channel blocker, FFA, would persist following catalase-induced H2O2 depletion. In their study, catalase alone caused a decrease in firing rate. However, when FFA (20 mM) was applied in the continued presence of catalase, they observed that the effects of FFA on firing was abolished, resulting in no change in firing rate from that observed with catalase alone. They concluded basal H2O2 is an important factor underlying the tonic activation of TRP channels. Cytosolic Ca2+ can be elevated by H2O2 (Citation40). This could lead to activation of a calcium-activated conductance such as that mediated by the TRPC3 channels, which are reported to be the sole TRP channels in SNr neurons of neonatal mouse (Citation47).
Role of catalase on TRPC5 and TRPC6 channels
A subtype of TRP channels that is relatively poorly understood are the TRPC (canonical) channels; which have the closest amino acid sequence to TRP (Citation13). Humans possess six TRPC-expressing genes, and mice contain seven. A TRPC protein common to both species is TRPC5 (Citation48). Early studies noted high expression of TRPC5 in the brain, but it was subsequently found in many, but not all, cells. TRPC5 has a key role in cells, including neuronal cells. A physiological stimulator has not been identified; instead there are multiple nonspecific stimulators, including receptor agonists (e.g. carbachol and ATP), endogenous lipids, and toxic metals. TRPC5 sensitivity to redox factors is shown by the effects of exogenous H2O2 and the redox protein thioredoxin (Citation49). Because of the selective effects on H2O2 and gadolinium chloride (Gd3+) responses, Naylor et al. (Citation50) investigated the hypothesis that Gd3+ responses depend on endogenous H2O2 production, using catalase to catalyze the conversion of H2O2 to water, vitamin C and gallic acid antioxidants and they observed that catalase inhibited H2O2 and the Gd3+ responses but not endogenous lipid (lysophosphatidylcholine) responses. They concluded that gallic acid and vitamin C suppress only TRPC5 activities that depend on H2O2, and lack effects on other nodes of channel activity.
Graham et al. (Citation51) investigated the underlying mechanism, particularly the roles of ROS and protein kinase C, in the diabetes-induced TRPC6 down regulation and they found that high glucose concentrations reduced TRPC6 protein expression in cultured mesangial cells. TRPC6 protein was also reduced in the glomeruli but not in the heart and aorta isolated from streptozotocin-induced diabetic rats. Catalase and superoxide dismutase enzymes prevented the inhibitory effects of high glucose on TRPC6.
Role of catalase on TRP polycystin- 2 (TRPP2)
In physiological terms, ROS have an inhibiting function of TRPP2 and it may play an important regulatory role by preventing a Ca2+ overload in stressed cells in placenta. In a study by Montalbetti et al. (Citation52), pre-incubation of human syncytiotrophoblast (hST) apical membranes with catalase (167 μg/mL) prevented increased lipid peroxidation on exposure to H2O2 (625 μM) for two hours. This is in somewhat agreement with the effect of H2O2 on hST TRPP2 channel function as opposed to the effect of purified lipids.
Role of catalase on TRPV1 channel
Nerve growth factor regulates the nociceptive properties of a subset of small diameter sensory neurons by increasing the expression of the heat-sensing TRP channel, TRPV1. This action involves activation of the tyrosine kinase receptor A/p38 MAPK pathway. Recent studies indicate that activation of tyrosine kinase receptor A promotes superoxide generation via NADPH oxidase. Puntambekar et al. (Citation53) determined whether the NADPH oxidase pathway is involved in nerve growth factor-stimulated TRPV1 expression using a rat pheochromocytoma 12 line and rat dorsal root ganglion neurons. In their study, treatment of these cells with NGF (100 ng/mL) increased TRPV1 protein expression (approx. two fold) but not mRNA. This increase was mimicked by H2O2 and attenuated by catalase (200 units/mL) and by inhibitors of NADPH oxidase. They concluded that NGF-mediated ROS generation in the induction of TRPV1 expression.
Conclusions
The results of recent studies indicated that mitochondrial depolarization and permeability transition in neuronal cells such as glia neuron are a main cause of reactive free oxygen radical production and loss of neuronal viability induced by complex I, II and III although catalase induces modulator effect on reactive free oxygen radical production in mitochondria due to its strong antioxidant specie. Recent studies reveal also that some TRP channels such as TRPM2, TRPV1 and TRPC5 free oxygen radical sensitive features of the TRP channels, which include stimulation by endogenous and exogenous H2O2 and relatively potent inhibition by the important antioxidant enzyme catalase. In addition, the identification of TRP as a key component of the neurological Ca2+ entry pathway in response to reactive oxygen species sheds new light on the physiology and pathophysiology of the neurological cells and brain. The reports suggest that catalase may act in part by suppressing oxidative stress-activated TRP channel activities, thus potentially suppressing unwanted cellular remodeling. Because there is substantial evidence for the deteriorating role of oxidative stress in neurological and brain dysfunction, manipulating mitochondrial and TRP function in the neurological cells by catalase may be highly useful in the future for experimental therapies of brain and neurological dysfunctions.
Acknowledgement
The author thanks to Mehmet Aykur (PhD student, Department of Biophysics) for preparing figures of the manuscript and Dr Peter Butterworth of King’s College London for assistance with the English language. There is no financial support in the current study.
Declaration of interest
The authors report no declarations of interest.
References
- Butterfield DA. Amyloid β-peptide [1-42]-associated free radical-induced oxidative stress and neurodegeneration in Alzheimer’s disease brain: mechanisms and consequences. Curr Med Chem 2003, 10, 2651–2659.
- Nazıroğlu M. Molecular Mechanisms of vitamin E on intracellular signaling pathways in brain. In Reactive oxygen species and diseases. Ed.Laszlo; Goth, Kerala, India: Research Signpost Press: 2007, pp 239–256.
- Halliwell B.Oxidative stress and neurodegeneration: where are we now? J Neurochem 2006,97,1634–1658.
- Naziroglu M. New molecular mechanisms on the activation of TRPM2 channels by oxidative stress and ADP-ribose. Neurochem Res 2007, 32, 1990–2001.
- Bond CE, Greenfield SA. Multiple cascade effects of oxidative stress on astroglia. Glia 2007, 55, 1348–1361.
- Dringen R. Oxidative and antioxidative potential of brain microglial cells. Antioxid Redox Signal 2005, 7, 1223–1233.
- Wilson JX. Antioxidant defense of the brain: a role for astrocytes. Can J Physiol Pharmacol 1997, 75, 1149–1163.
- Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N, Kurose H, Okada Y, Imoto K, Mori Y. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell 2002, 9, 163–173.
- Wehage E, Eisfeld J, Heiner I, Jüngling E, Zitt C, Lückhoff A. Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J Biol Chem 2002, 277, 23150–23156.
- Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev 2007, 87, 165–217.
- Naziroglu M. TRPM2 cation channels, oxidative stress and neurological diseases: where are we now? Neurochem Res 2011, 36, 355–366.
- Heiner I, Eisfeld J, Warnstedt M, Radukina N, Jüngling E, Lückhoff A. Endogenous ADP-ribose enables calcium-regulated cation currents through TRPM2 channels in neutrophil granulocytes. Biochem J 2006, 398, 225–232.
- Putney JW, Smyth JT, Trebak M, Lemonnier L, Vazquez G, Gary S, Bird GS. Activation and regulation of TRPC cation channels. Cell Membr Free Radic Res 2008, 1, 51–55.
- Toescu EC, Vreugdenhil M. Calcium and normal brain ageing. Cell Calcium 2010, 47, 158–164.
- Pariente JA, Camello C, Camello PJ, Salido GM. Release of calcium from mitochondrial and nonmitochondrial intracellular stores in mouse pancreatic acinar cells by hydrogen peroxide. J Membr Biol 2001, 179, 27–35.
- Bejarano I, Terrón MP, Paredes SD, Barriga C, Rodríguez AB, Pariente JA. Hydrogen peroxide increases the phagocytic function of human neutrophils by calcium mobilisation. Mol Cell Biochem 2007, 296, 77–84.
- Campanucci VA, Krishnaswamy A, Cooper E. Mitochondrial reactive oxygen species inactivate neuronal nicotinic acetylcholine receptors and induce long-term depression of fast nicotinic synaptic transmission. J Neurosci 2008, 28, 1733–1744.
- Yokoyama H, Uchida H, Kuroiwa H, Kasahara J, Araki T. Role of glial cells in neurotoxin-induced animal models of Parkinson’s disease. Neurol Sci 2011, 32, 1–7.
- Bélanger M, Magistretti PJ. The role of astroglia in neuroprotection. Dialogues Clin Neurosci 2009, 11, 281–295.
- Abramov AY, Jacobson J, Wientjes F, Hothersall J, Canevari L, Duchen MR. Expression and modulation of an NADPH oxidase in mammalian astrocytes. J Neurosci 2005, 25, 9176–9184.
- Safiulina VF, Afzalov R, Khiroug L, Cherubini E, Giniatullin R. Reactive oxygen species mediate the potentiating effects of ATP on GABAergic synaptic transmission in the immature hippocampus. J Biol Chem 2006, 281, 23464–23470.
- Jenner P, Olanow CW. Oxidative stress and the pathogenesis of Parkinson’s disease. Neurology 1996, 47, S161–S170.
- Hajnóczky G, Csordás G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, Yi M. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 2006, 40, 553–560.
- Hwang SL, Yen GC. Neuroprotective effects of the citrus flavanones against H2O2-induced cytotoxicity in PC12 cells. J Agric Food Chem 2008, 56, 859–864.
- Ozgül C, Naziroglu M. TRPM2 channel protective properties of N-acetylcysteine on cytosolic glutathione depletion dependent oxidative stress and Ca(2+) influx in rat dorsal root ganglion. Physiol Behav 2012, 106, 122–128.
- Bejarano I, Redondo PC, Espino J, Rosado JA, Paredes SD, Barriga C, Reiter RJ, Pariente JA, Rodríguez AB. Melatonin induces mitochondrial-mediated apoptosis in human myeloid HL-60 cells. J Pineal Res 2009, 46, 392–400.
- Espino J, Bejarano I, Paredes SD, Barriga C, Reiter RJ, Pariente JA, Rodríguez AB. Melatonin is able to delay endoplasmic reticulum stress-induced apoptosis in leukocytes from elderly humans. Age (Dordr) 2011, 33, 497–507.
- Kowaltowski AJ, Castilho RF, Vercesi AE. Opening of the mitochondrial permeability transition pore by uncoupling or inorganic phosphate in the presence of Ca2+ is dependent on mitochondrial-generated reactive oxygen species. FEBS Lett 1996, 378, 150–152.
- Maciel EN, Kowaltowski AJ, Schwalm FD, Rodrigues JM, Souza DO, Vercesi AE, Wajner M, Castilho RF. Mitochondrial permeability transition in neuronal damage promoted by Ca2+ and respiratory chain complex II inhibition. J Neurochem 2004, 90, 1025–1035.
- Vesce S, Kirk L, Nicholls DG. Relationships between superoxide levels and delayed calcium deregulation in cultured cerebellar granule cells exposed continuously to glutamate. J Neurochem 2004, 90, 683–693.
- Clapham DE. SnapShot: mammalian TRP channels. Cell 2007, 129, 220.
- Perraud AL, Takanishi CL, Shen B, Kang S, Smith MK, Schmitz C, Knowles HM, Ferraris D, Li W, Zhang J, Stoddard BL, Scharenberg AM. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J Biol Chem 2005, 280, 6138–6148.
- Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, Kinet JP, Scharenberg AM. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001, 411, 595–599.
- Olah ME, Jackson MF, Li H, Perez Y, Sun HS, Kiyonaka S, Mori Y, Tymianski M, MacDonald JF. Ca2+-dependent induction of TRPM2 currents in hippocampal neurons. J Physiol (Lond) 2009, 587, 965–979.
- Xie YF, Belrose JC, Lei G, Tymianski M, Mori Y, Macdonald JF, Jackson MF. Dependence of NMDA/GSK-3ß Mediated Metaplasticity on TRPM2 Channels at Hippocampal CA3-CA1 Synapses. Mol Brain 2011, 4, 44.
- Naziroglu M, Özgül C, Çelik Ö, Çig B, Sözbir E. Aminoethoxydiphenyl borate and flufenamic acid inhibit Ca2+ influx through TRPM2 channels in rat dorsal root ganglion neurons activated by ADP-ribose and rotenone. J Membr Biol 2011, 241, 69–75.
- Naziroglu M, Lückhoff A, Jüngling E. Antagonist effect of flufenamic acid on TRPM2 cation channels activated by hydrogen peroxide. Cell Biochem Funct 2007, 25, 383–387.
- Bové J, Prou D, Perier C, Przedborski S. Toxin-induced models of Parkinson’s disease. NeuroRx 2005, 2, 484–494.
- Cormier A, Morin C, Zini R, Tillement JP, Lagrue G. Nicotine protects rat brain mitochondria against experimental injuries. Neuropharmacology 2003, 44, 642–652.
- Freestone PS, Chung KK, Guatteo E, Mercuri NB, Nicholson LF, Lipski J. Acute action of rotenone on nigral dopaminergic neurons–involvement of reactive oxygen species and disruption of Ca2+ homeostasis. Eur J Neurosci 2009, 30, 1849–1859.
- Nilius B. TRP channels in disease. Biochim Biophys Acta 2007, 1772, 805–812.
- Bao L, Avshalumov MV, Rice ME. Partial mitochondrial inhibition causes striatal dopamine release suppression and medium spiny neuron depolarization via H2O2 elevation, not ATP depletion. J Neurosci 2005, 25, 10029–10040.
- Selvaraj S, Sun Y, Singh BB. TRPC channels and their implication in neurological diseases. CNS Neurol Disord Drug Targets 2010, 9, 94–104.
- Balzer M, Lintschinger B, Groschner K. Evidence for a role of Trp proteins in the oxidative stress-induced membrane conductances of porcine aortic endothelial cells. Cardiovasc Res 1999, 42, 543–549.
- Poteser M, Graziani A, Rosker C, Eder P, Derler I, Kahr H, Zhu MX, Romanin C, Groschner K. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J Biol Chem 2006, 281, 13588–13595.
- Lee CR, Witkovsky P, Rice ME. Regulation of Substantia Nigra Pars Reticulata GABAergic Neuron Activity by H2O2 via Flufenamic Acid-Sensitive Channels and K(ATP) Channels. Front Syst Neurosci 2011, 5, 14.
- Zhou FW, Matta SG, Zhou FM. Constitutively active TRPC3 channels regulate basal ganglia output neurons. J Neurosci 2008, 28, 473–482.
- Clapham DE. TRP channels as cellular sensors. Nature 2003, 426, 517–524.
- Xu C, Macciardi F, Li PP, Yoon IS, Cooke RG, Hughes B, Parikh SV, McIntyre RS, Kennedy JL, Warsh JJ. Association of the putative susceptibility gene, transient receptor potential protein melastatin type 2, with bipolar disorder. Am J Med Genet B Neuropsychiatr Genet 2006, 141B, 36–43.
- Naylor J, Al-Shawaf E, McKeown L, Manna PT, Porter KE, O’Regan D, Muraki K, Beech DJ. TRPC5 channel sensitivities to antioxidants and hydroxylated stilbenes. J Biol Chem 2011, 286, 5078–5086.
- Graham S, Gorin Y, Abboud HE, Ding M, Lee DY, Shi H, Ding Y, Ma R. Abundance of TRPC6 protein in glomerular mesangial cells is decreased by ROS and PKC in diabetes. Am J Physiol, Cell Physiol 2011, 301, C304–C315.
- Montalbetti N, Cantero MR, Dalghi MG, Cantiello HF. Reactive oxygen species inhibit polycystin-2 (TRPP2) cation channel activity in term human syncytiotrophoblast. Placenta 2008, 29, 510–518.
- Puntambekar P, Mukherjea D, Jajoo S, Ramkumar V. Essential role of Rac1/NADPH oxidase in nerve growth factor induction of TRPV1 expression. J Neurochem 2005, 95, 1689–1703.