
Abstract
Purpose: Congenital hereditary endothelial dystrophy (CHED) is a rare genetic disorder caused by mutations in corneal endothelial cells. CHED can be divided into 2 types by the modes of inheritance; CHED type 1 (CHED1) with autosomal dominant inheritance and CHED type 2 (CHED2) with autosomal recessive inheritance. Mutations in the sodium bicarbonate transporter-like solute carrier family 4 member 11 (SLC4A11) gene result CHED2.
Methods: A 37 years old female was clinically diagnosed as CHED2. Peripheral blood from the patient and her family members was obtained under informed consents. Genomic DNA was extracted in their WBCs, and whole exons and exon-intron boundaries of the SLC4A11 gene were amplified using polymerase chain reaction. The amplified materials were analyzed by direct sequencing method.
Results: The sequencing results of the SLC4A11 gene showed a novel homozygous mutation in exon 9 (c.1158C > A, p.C386*) in the proband with CHED2 phenotype. Her father and sister showing normal cornea were heterozygous carriers of the mutation. Her mother showing late onset Fuchs endothelial corneal dystrophy (FECD) also had the same mutation heterozygously.
Discussion: We report a novel nonsense mutation of the SLC4A11 gene in the patient with CHED2. In addition, one of heterozygous carriers in this family showed features of late onset FECD. Close clinical ocular examination for the heterozygous carriers should be performed to detect late onset FECD.
Congenital hereditary endothelial dystrophy (CHED) is a rare genetic disorder caused by mutations in genes associated with corneal endothelial cells, which is characterized by bilateral symmetric corneal opacity and edema. CHED can be divided into two types by the modes of inheritance; CHED type 1 (CHED1, MIM#121700) with autosomal dominant inheritance and CHED type 2 (CHED2, MIM#217700) with autosomal recessive inheritance.Citation1 Among them, the sodium bicarbonate transporter-like solute carrier family 4 member 11 (SLC4A11) gene (MIM#610206) has been identified as a gene which causes CHED2.Citation1–5
The SLC4A11 gene is located at chromosome 20p12 and encodes 891 amino acids membrane protein, reported as a sodium-coupled borate cotransporter 1 and highly expressed in the corneal endothelium. It has been known that mutations of this gene causes three corneal dystrophies; CHED2, CDPD, and Fuchs endothelial corneal dystrophy (FECD).Citation6 In particular, heterozygous SLC4A11 mutations could cause late onset FECD, and late onset FECD might occur in the parents of CHED2 patients, who are heterozygous carriers of SLC4A11 mutations.Citation7
We recently identified a novel mutation of the SLC4A11 gene found in a patient with CHED2 and one of the parents with late onset FECD. Here, we report these cases to emphasize the possibility of late onset corneal diseases in the CHED2 patients’ parents who have the mutation heterozygously and the necessity of close examinations for the family members of CHED2 patients.
A 37-year-old woman visited our hospital with low visual acuity and known corneal opacity in both eyes, originating from early childhood. She did not have any photophobia, nystagmus, strabismus or hearing difficulty. Her parents were not consanguineous, and she said that none of her family members had ocular symptoms. Her uncorrected visual acuity (UCVA) was 20/80 in both eyes and the manifest refraction was uncorrectable. Intraocular pressure was 15 mmHg in the right eye and 14 mmHg in the left. Slit-lamp examination revealed bilateral corneal haze over the entire corneas, with diffuse stromal edema. Ultrasonic pachymetry (Alcon-Surgical, Inc., Irvine, CA) yielded values of 1036 µm and 1033 µm for central corneal thickness in the right and left cornea, respectively. Specular microscopic evaluation (Konan Noncon Robo SP 8000; Konan Medical, Inc., Hyogo, Japan) could not be performed owing to corneal edema. Confocal microscopy (ConfoScan 4; Nidek, Gamagori, Japan) demonstrated increased corneal thickness and 995 cell/mmCitation2 of endothelial cell density. CHED2 was suspected, and molecular genetic analysis for CHED2 was performed. Ocular examinations were recommended to her relatives and performed.
The proband’s father and sister were clinically unaffected. The proband’s 62-year-old mother was also screened for corneal diseases as a family member of the CHED2 patient. She had no subjective symptoms. Her best corrected visual acuity (BCVA) was 20/20 and her refraction was +1.75 diopter (D) +1.00 D × 165° in the right eye. In the left eye, the BCVA was 20/20 and refraction was +1.25 D + 1.00 D × 180°. However, slit-lamp examination and specular microscopic evaluation revealed bilateral corneal guttatae over the entire corneas and mild cataract on both her eyes. Corneal thickness was 613 μm in her right eye and 604 μm in her left eye. Late onset FECD was suspected, and all coding exons of SLC4A11 analysis were also performed.
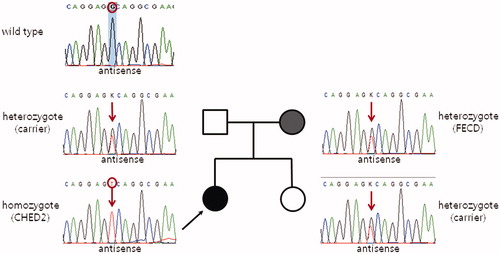
Results of DNA sequencing revealed that the proband was homozygous for a novel nonsense mutation (c.1158C > A) in exon 9, which caused premature termination of the SLC4A11 protein (p.Cys386*, ). Her 64-year-old father, mother (mentioned above), and 36-year-old sister also harbored this mutation heterozygously ().
FIGURE 1. Partial sequences of the SLC4A11 gene show the mutation detected in this study. A novel nonsense mutation in exon 9 (c.1158C > A) leads to premature termination of the SLC4A11 protein instead of the 386th cysteine (p.C386*). The proband’s mother (Case 2), a heterozygous mutation carrier, showed late onset Fuchs endothelial corneal dystrophy phenotypes. Other family members with the mutation heterozygously had no corneal diseases clinically.
Though another missense mutation at the same amino acid location (c.1156T > C, p.Cys386Arg) was already reported, the mutation identified in our study was novel located in the different nucleotide site and led to the different SLC4A11 mutant.Citation3 Also, the location of this mutation was in the first transmembrane segment (p.Tyr375 to p.Leu396) among 14 segments of the transmembrane region, where disease-causing mutations were predominantly found.Citation8,Citation9 Therefore, we postulated that the fore location of the mutation found in our study caused premature termination of the SLC4A11 protein and led to the failure to locate in the membrane and intracellular retention.
The SLC4A11 dimerization had a role in the onset age of symptoms and dominant versus recessive inheritance of the corneal dystrophies.Citation9 As mentioned above, we revealed that the proband with CHED2 was homozygote of p.Cys386* and all family members were heterozygous carrier of the mutation. Furthermore, the mother of the proband, a carrier with p.Cys386*, was clinically diagnosed with late onset FECD. Our cases confirm that SLC4A11 mutations could cause recessive CHED2. We suggest that close ocular examination of family members should be performed.Citation7 Because the diagnosis of FECD for the mother was not based on pathological findings, there was also a possibility that another mutation in a different gene is associated with late onset FECD.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Aldave AJ, Yellore VS, Bourla N, et al. Autosomal recessive CHED associated with novel compound heterozygous mutations in SLC4A11. Cornea 2007;26:896–900
- Jiao X, Sultana A, Garg P, et al. Autosomal recessive corneal endothelial dystrophy (CHED2) is associated with mutations in SLC4A11. J Med Genet 2007;44:64–68
- Ramprasad VL, Ebenezer ND, Aung T, et al. Novel SLC4A11 mutations in patients with recessive congenital hereditary endothelial dystrophy (CHED2). Mutation in brief #958. Online. Hum Mutat 2007;28:522–523
- Hemadevi B, Veitia RA, Srinivasan M, et al. Identification of mutations in the SLC4A11 gene in patients with recessive congenital hereditary endothelial dystrophy. Arch Ophthalmol 2008;126:700–708
- Sultana A, Garg P, Ramamurthy B, et al. Mutational spectrum of the SLC4A11 gene in autosomal recessive congenital hereditary endothelial dystrophy. Mol Vis 2007;13:1327–1332
- Vilas GL, Morgan PE, Loganathan SK, et al. A biochemical framework for SLC4A11, the plasma membrane protein defective in corneal dystrophies. Biochemistry 2011;50:2157–2169
- Vithana EN, Morgan PE, Ramprasad V, et al. SLC4A11 mutations in Fuchs endothelial corneal dystrophy. Hum Mol Genet 2008;17:656–666
- Vithana EN, Morgan P, Sundaresan P, et al. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2). Nat Genet 2006;38:755–757
- Vilas GL, Loganathan SK, Quon A, et al. Oligomerization of SLC4A11 protein and the severity of FECD and CHED2 corneal dystrophies caused by SLC4A11 mutations. Hum Mutat 2012;33:419–428