Abstract
Context: We recently reported that F2, an oligomer procyanidin fraction isolated from grape seeds, triggered an original form of cell death in U-87 human glioblastoma cells with a phenotype resembling morphological characteristics of paraptosis. However, the specific death mode induced by F2 and the mechanism of its action have not been assessed so far.
Objective: In the present work, we therefore further investigated the death mode of human glioblastma cells induced by F2 and gained insight into the nature of the signaling pathways activated by F2 in glioblastoma cells.
Materials and methods: Cell viability assay using MTT, (AO/EB) double staining, Western blot analysis, and Ca2+ assay using fura-2.
Results: Morphology studies revealed extensive cytoplasmic vacuolization in dying cells and no apoptotic body formation, membrane bleb formation, or nuclear fragmentation, though some was accompanied by MAPK activation and new protein synthesis, and was independent of caspase activation. Moreover, we demonstrated the involvement of calcium mobilization in F2-induced U-87 cell signaling.
Discussion and conclusion: Altogether we showed that F2 induced a kind of cell death resembling paraptosis in U-87 cells. The current report complements previous studies on the characterization of F2-induced U-87 cell death, enhances our understanding of the action mechanism of F2 on glioma, and helps in the development of novel antitumor therapeutics.
Introduction
In recent years, it has become evident that the classic dichotomy of apoptosis versus necrosis is too simple to encompass the wide spectrum of cell death possibilities to guard the organism against unwanted and potentially harmful cells, and more and more evidence suggests that cell death may be classified into two main types: programmed cell death (PCD), in which the cell plays an active role in its own demise, and passive (necrotic) cell death. The PCD observed during development and tissue homeostasis has been classified morphologically into three main types: type 1, also known as nuclear or apoptotic; type 2 or autophagic; and type 3, also referred to as paraptosis (CitationClarke, 1990).
Although PCD has often been equated with apoptosis and apoptosis is the best-characterized type of PCD, there is increasing evidence that the alternative, non-apoptotic PCD exists in parallel with apoptosis. Recently, paraptosis, a distinct non-apoptotic variant of PCD in multicellular organisms, has been paid more attention (CitationAsare et al., 2008; CitationSperandio et al., 2000; CitationValamanesh et al., 2007). Paraptotic cells are characterized by a swelling and vacuolization process that begins with progressive swelling of mitochondria and the endoplasmic reticulum (ER), condensation of chromatin (but not early margination of the chromatin), and requiring RNA and protein synthesis, yet characteristics of apoptosis-like cell shrinkage, cell surface blebbing, the formation of apoptotic bodies, and activation of caspases do not appear (CitationSperandio et al., 2000). Moreover, paraptosis has been described as mediated by mitogen-activated protein kinases (CitationSperandio et al., 2004), and can be triggered by the tumor necrosis factor (TNF) receptor family member TAJ/TROY (CitationWang et al., 2004) and neurokinin-1 receptor (CitationCastro-Obregon et al., 2002), as well as the insulin-like growth factor I receptor (IGFR-I) (CitationSperandio et al., 2000). Furthermore, T9 glioma cells expressing membrane macrophage colony-stimulating factor were killed by polymorphonuclear leukocytes and macrophages with vacuolization of the mitochondria and ER, resembling paraptosis (CitationChen et al., 2002).
Successful treatment with chemotherapeutic drugs is largely dependent on their ability to trigger cell death in tumor cells, and the majority of cytotoxic agents trigger the apoptosis pathway. However, evidence suggests that there are forms of chemotherapy-induced cell death that cannot readily be classified as apoptosis or necrosis but fit more in the paraptosis PCD model (CitationLeist & Jaattela, 2001). In previous studies, we have observed that the grape seed extract F2 may induce a form of non-apoptotic PCD, dubbed paraptosis, in human glioblastoma U-87 cells (CitationZhang et al., 2009). Therefore, it is interesting to further investigate the F2-induced death mode in U-87 cells and gain an insight into the nature of its signaling pathways.
Materials and methods
Cell culture and reagents
Human glioblastoma cell line U-87 (American Type Culture Collection, ATCC) was grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS), 100 μg/mL streptomycin, and 100 IU/mL penicillin in a humidified atmosphere of 95% air and 5% CO2. Antibodies to both procaspase-3 and caspase-3 were purchased from Santa Cruz (USA); Antibodies to both phosphorylated and total ERK (extracellular signal-regulated kinase) as well as p38 were purchased from Cell Signaling Technology (USA). All the other reagents were purchased from Sigma (St Louis, MO, USA) if not otherwise stated.
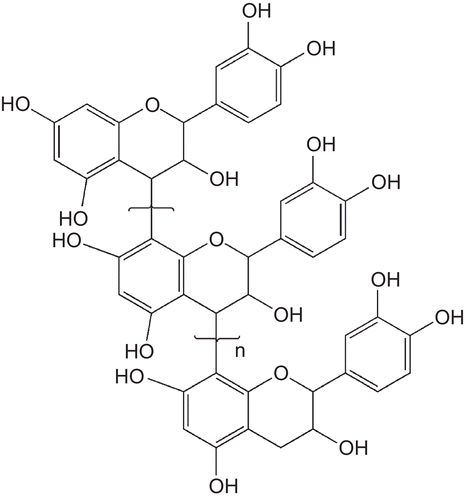
F2 is an oligomer procyanidin from grape seeds (Vitis vinifera, cv. Fernao Pires) with flavan-3-ol monomer units (catechins) linked mostly by acid-labile 4→8 and, in some cases, by 4→6 bonds, as previously reported (CitationGuo et al., 2007; CitationSpranger et al., 2008; CitationSun et al., 1998, Citation1999) (). It was dissolved in pure water before use.
Figure 1. General chemical structure of oligomer procyanidins (n = 2–15).
Cell viability assay
The cell viability was measured using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Briefly, 5000 cells were seeded in each well of a 96-well culture plate. After 24 h, cells were treated with F2 at concentrations ranging from 0 to 100 μg/mL for 24 h. MTT was added and incubated for 3 h. The formed formazan was dissolved by dimethylsulfoxide (DMSO). The absorbance of each well was measured with a microplate reader at a wavelength of 492 nm. To calculate IC50, percent inhibitions were plotted as a function of log[concentration] and then fit to a four-parameter logistic model that allowed for a variable Hill slope by using XLFIT 4.1 (ID Business Solutions, Emeryville, CA).
Acridine orange/ethidium bromide (AO/EB) double staining
Dye mixture (4 μL, 100 μg/mL AO and 100 μg/mL EB in distilled water) was mixed with 100 μL phosphate buffered saline (PBS). Cells were seeded on a clean microscope slide. After treatment with F2, 30 μL dye mixture was dropped on the cell slide and immediately examined by fluorescence microscopy at ×400 magnification.
Western blot analysis
The ERK1/2 and p38 MAPK (mitogen-activated protein kinase) activities were determined by Western blotting using specific antibodies to both phosphorylated and total ERK1/2 and p38. Procaspase-3 expression and caspase-3 activation were assessed by Western blotting using their specific antibodies. Cells were preincubated with different concentrations of F2 for the indicated time points. Cell activation was terminated by placing the cells on ice, aspirating the medium, and immediately lysing the cells in RIPA (radioimmuno precipitation assay) lysis buffer. Total protein was determined followed by the addition of loading buffer and heat denaturation prior to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 12% gels. Separated proteins were electrotransferred to polyvinylidine fluoride (PVDF) membranes presoaked in transfer buffer at 300 mA per blot for 50 min. Membranes were blocked with 5% nonfat milk in Tris-buffered saline (pH 7.4) for 60 min, followed by overnight incubation at 4°C in the relevant primary antibody (1:1000), and finally detection for 1 h with a secondary antibody horseradish peroxidase (HRP)-conjugate (1:2000). Immunoreactive protein bands were detected by enhanced chemiluminescence.
Ca2+ assay
The intracellular Ca2+ concentrations were measured using fluorescent indicator fura-2. Ca2+ mobilization was measured by incubating 2 × 107 tumor cells in 1 mL of loading medium (DMEM containing 10% FBS and 2 mmol/L glutamine) with 10 μM fura-2 acetoxymethyl ester (Biotium) for 45 min at room temperature. The dye-loaded cells were washed and resuspended in saline buffer [138 mM NaCl, 6 mM KCl, 1 mM CaCl2, 10 mM HEPES, 5 mM glucose, and 0.1% bovine serum albumin (pH 7.4)] at a density of 5 × 106/mL. Cells were then transferred into 96-well microplates (1 × 106 cells in 200 μL of saline buffer), and the microplates were placed in a Synergy™ HT multi-detection microplate reader with excitation at 340 nm and 380 nm and emission at 500 nm. Stimulants were added to the wells in a volume of 10 μL, and the intensity of the fluorescence was measured by use of the ratio of absorbance at 340 to that at 380 nm, calculated with the formula described earlier (CitationHallett et al., 1999).
Statistical analysis
Results are expressed as mean ± SE of three experiments performed in triplicate samples. One-way analysis of variance (ANOVA) followed by Dunnett’s t-test was used for statistical analysis (SPSS 13.0 software; SPSS, USA).
Results
F2-induced programmed cell death in U-87 cells
Previous experiments indicated that F2 markedly inhibited the proliferation of U-87 cells, with an IC50 of 75 μg/mL at 24 h, 33.6 μg/mL at 48 h, and 25 μg/mL at 72 h. As shown in , F2 was a more potent antiglioma agent than 5-fluorouracil (5-Fu). Treatment with F2 at the concentration of 30 μg/mL over a 24 h period significantly inhibited glioblastoma cell viability in vitro as measured by MTT assay.
Figure 2. Effect of F2 on U-87 cell viability and the effect of cycloheximide (CHX) on F2-induced U-87 cell death. (A) Cells were treated with increasing concentrations of F2 or 5-Fu (as positive control) for 24 h. Results are expressed as percentage values taking control group as 100%. (B) Effect of CHX on F2-induced U-87 cell death. Cells were exposed to F2 (30 μg/mL) for 24 h with or without CHX (0.1 μg/mL) and analyzed by MTT assay. Data herein are represented as mean ± SE of three separate experiments. **p < 0.01, ***p < 0.001 compared to control group, #p < 0.05 statistically significant difference from F2-treated cells and F2-treated cells in combination with CHX, Dunnett’s test.
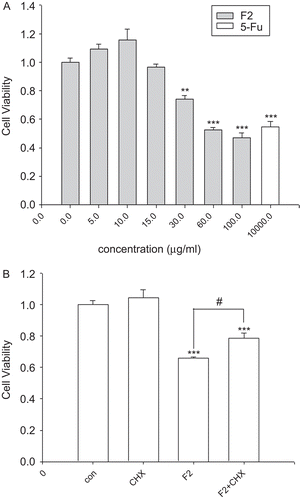
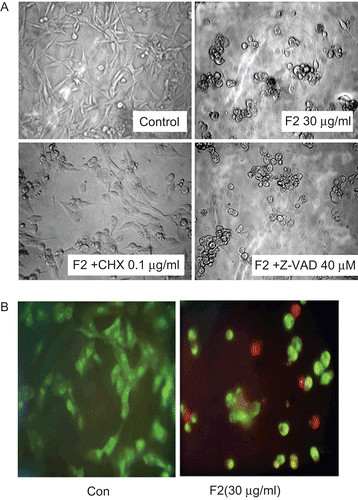
To investigate the possibility of a passive cell death, for example necrosis, cycloheximide (CHX) was employed to block de novo protein synthesis (CitationGardner et al., 1998). Cells were exposed to CHX (0.1 μg/mL) in combination with F2 (30 μg/mL) and a significant inhibition of cell death was observed. Indeed, the cell viability percentage observed with F2 treatment was 65.9%, increasing to 78.6% when CHX was used (). An even more pronounced effect of CHX was observed on F2-induced vacuolization. As shown in , CHX markedly reduced the cytoplasmic vacuolization, suggesting that F2-induced U-87 cell death requires protein synthesis. In addition, it is reported that the passive necrotic cells show a loss of plasma membrane integrity, swelling, and then lysis (CitationWyllie et al., 1980), whereas in most of the cells treated with F2, destruction of the plasmic membrane was not observed (). Thus, it is suggested that F2 induces programmed cell death in U-87 cells.
Figure 3. Effect of F2 on observation of microscopy changes in U-87 cells. (A) Effect of cycloheximide/Z-VAD-FMK on F2-induced cell vacuolization. Cells were exposed to F2 (30 μg/mL) for 24 h with or without cycloheximide (CHX; 0.1 μg/mL) or Z-VAD-FMK (Z-VAD; 40 μM), and analyzed by light microscopy. (B) Fluorescence microscopy changes in U-87 cells. Cells were treated with or without F2 (30 μg/mL) for 24 h and AO/EB staining was performed. AO is taken up by both viable and nonviable cells and emits green fluorescence if intercalated into DNA. EB is taken up only by nonviable cells and emits red fluorescence by intercalation into DNA. Results are from a single experiment; similar results were obtained in three separate experiments.
F2-induced non-apoptotic cell death in U-87 cells
Cells were exposed to F2 over a 24 h period and examined by microscopy. Images in clearly reveal that F2 caused a dramatic increase in vacuolization upon exposure, and the cells tended to round up. There were no signs of classic apoptotic changes, such as cell shrinkage or membrane blebbing. Fluorescence microscopic studies of cells stained with AO/EB also confirmed that the cells evolved in a sequence in which chromatin condensation and reduction of nucleus size occurred together with cytoplasmic vacuolization (). Taken together, the morphologic changes induced by F2 were mostly suggestive of paraptosis.
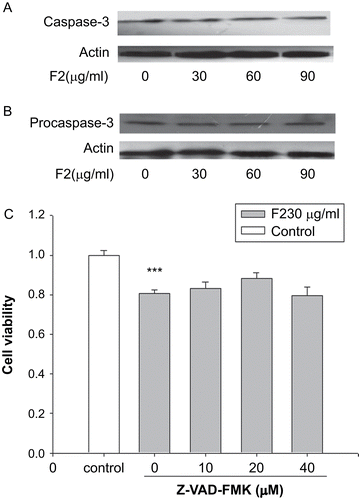
Since caspases are considered to be the core effectors of apoptosis and caspase-3 is a key mediator of both receptor and mitochondria mediated apoptosis, F2 was tested for its ability to induce caspase-3 activation in lysates of U-87 cells (). Treatment with 30, 60, and 90 μg/mL F2 did not result in a significant activation of caspase-3. To further demonstrate the absence of caspase-3 activation in F2-treated cells, we performed Western blot by using an antibody recognizing procaspase-3. Indeed, exposure to F2 did not alter the expression level of procaspase-3 in comparison to untreated cells (). In addition, as illustrated in , pan-caspase inhibitor Z-VAD-FMK (10–40 μM) could not attenuate F2-induced U-87 cell death. Similar findings were observed in that up to 40 μM, Z-VAD-FMK had no apparent effect on the observed cytoplasmic vacuolization induced by F2 treatment (). Thus, these results, pointing out a scarce presence of classical signs of apoptosis, suggested that F2-treated cells mainly died through a non-apoptotic cell death mechanism.
Figure 4. F2-induced non-apoptotic cell death in U-87 cells. (A) Effect of F2 on caspase-3 activation. Cells were preincubated with 30, 60, and 90 μg/mL F2 for 12 h. The cell lysates were examined for caspase-3 activation by Western blotting. Actin proteins were examined as a loading control to demonstrate the levels of protein loading. (B) Effect of F2 on procaspase-3 expression in U-87 cells. Cells were preincubated with 30, 60, and 90 μg/mL F2 for 12 h. The cell lysates were examined for procaspase-3 expression by Western blotting. Actin proteins were examined as a loading control to demonstrate the levels of protein loading. (C) Effect of Z-VAD-FMK (Z-VAD) on F2-induced U-87 cell death. Cells were exposed to F2 (30 μg/mL) for 24 h with or without Z-VAD and analyzed by MTT assay. Data herein are represented as mean ± SE of three separate experiments. ***p < 0.001 compared to control group, Dunnett’s test.
F2-induced cell death lacked the specific markers for autophagy
As the presence of multiple vacuoles may suggest activation of autophagy (CitationBursch et al., 2000), an experiment was performed to analyze autophagy using monodansylcadaverin (MDC), a protein of the autophagic vesicles. It was found that MDC labeling did not significantly differ in F2-treated and control cells (data not shown), suggesting that F2-induced cell death lacked the specific markers for autophagy.
F2-induced paraptosis-like cell death by cytoplasm vacuolization
We showed F2-induced U-87 cell death to be distinct from apoptosis, necrosis, or autophagy. Morphology studies revealed that cells tended to be round-shaped, with extensive cytoplasmic vacuolization after treatment with F2 (). Indeed, the death resembled the central characteristic of paraptosis, and F2 induced an alternative programmed cell death resembling paraptosis in U-87 cells.
Nature of signaling pathways activated by F2
F2-triggered activation of ERK1/2 and p38 in U-87 cells
The MAPKs have been suggested to play important roles in paraptosis (CitationSperandio et al., 2004). Accordingly, as illustrated from the Western analyses presented in and B, marked activations of ERK1/2 and p38 were observed, as shown by increased levels of their phosphorylated forms after 6 and 12 h of treatment. Thus, F2 triggered the activation of ERK1/2 and p38 in U-87 cells.
Figure 5. Signal pathways activated by F2 in U-87 cells. (A) Effect of F2 on phosphorylation of ERK1/2. Cells were preincubated with 30 and 60 μg/mL F2 for 6 and 12 h, respectively. Cell lysates were examined for ERK1/2 activation by Western blotting. Total ERK1/2 proteins were examined as a loading control to demonstrate the levels of protein loading. (B) Effect of F2 on phosphorylation of p38. Cells were preincubated with 30 and 60 μg/mL F2 for 6 h and 12 h, respectively. Cell lysates were examined for p38 activation by Western blotting. Total p38 proteins were examined as a loading control to demonstrate the levels of protein loading. (C) Effect of F2 on U-87 cell calcium mobilization. (a) F2-induced Ca2+ flux of U-87 cells. (b) The main source for F2-induced Ca2+ release and the effect of PTX on its release. Cells loaded with fura-2 were triggered with F2 (100 μg/mL) in the absence of extracellular Ca2+ (no Ca2+ was added to the buffer, which was supplemented with 0.5 mM EGTA) or cells were pretreated with 100 ng/mL PTX for 16 h then F2 (100 μg/mL) was added. The intensity of fluorescence was measured by use of the ratio of absorbance at 340 to that at 380 nm.
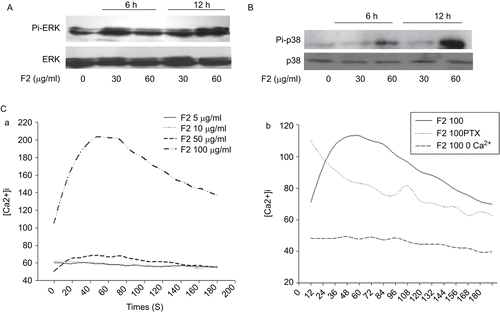
F2-triggered calcium mobilization in U-87 cells
Calcium plays an important role in physiological cell death processes. Cell injury occurs when the intracellular calcium pool is disturbed, which in turn may lead to cell death (CitationGupta & Pushkala, 1999). We then focused on the measurement of calcium mobilization to test whether F2 was able to elicit Ca2+ flux in U-87 cells. As shown in , at lower concentrations, F2 lacked significant Ca2+ mobilizing activity in U-87 cells; however, at a concentration of 50 μg/mL it exhibited a slight Ca2+ flux, and at a concentration of 100 μg/mL it promoted significant intracellular Ca2+ flux in U-87 cells. In order to investigate the source of Ca2+ release in response to F2, cells were challenged with F2 in the absence of extracellular Ca2+ (no Ca2+ was added to the buffer, which was supplemented with 0.5 mM ethylene glycol tetraacetic acid (EGTA)). It was found that the F2-induced increase in [Ca2+]i was mainly due to the influx of extracellular Ca2+. In addition, pre-treatment with pertussis toxin (PTX) also caused inhibition in the F2-induced Ca2+ response, attesting to the participation of some Gi/Go proteins in F2-induced U-87 cell calcium mobilization ().
Disscussion
Malignant glioma is the most common primary malignant tumor within the central nervous system. Even though there has been progression in treatments including surgery, chemotherapy, and radiotherapy, the median survival time of patients with malignant glioma is less than 1 year from diagnosis. Therefore, there is an urgent need to develop a new potential antiglioma compound to optimize the antitumor effect. It is reported that procyanidins isolated from grape seeds have some activities in inhibiting tumor growth (CitationYe et al., 1999). Previous observations by our laboratory showed that F2, an oligomeric procyanidin fraction of grape seeds, could inhibit the cell viability of human malignant glioma U-87 cells (CitationZhang et al., 2009). However, the action mechanism of F2 for inducing U-87 cell death is not clear.
It has been demonstrated in the present study that F2-induced glioblastoma cell death is distinct from apoptosis, necrosis, or autophagy. Upon exposure to F2, a dramatic increase in cytoplasmic vacuolization was observed. There is increasing evidence that cytoplasmic vacuolization is considered to be a central characteristic of paraptosis, and vacuolization is supposed to be caused by ionic disregulation accompanied by water influx and retention, which lead to physical swelling of the mitochondria and endoplasmic reticulum (ER) (CitationHoa et al., 2007). Accordingly, in our previous study, F2 was found to induce an early and very marked cytoplasmic vacuolization, most probably caused by severe perturbations in mitochondrial membrane potential. In addition to cytoplasmic vacuolization, other features, typical characteristics of paraptotic cells, were also found in the study. The death process demonstrated no apoptotic body formation, membrane bleb formation, and nuclear fragmentation, though some degree of chromatin condensation occurred. Furthermore, F2-induced U-87 cell death was independent of caspase activation (pan-caspase inhibitor Z-VAD-FMK could not attenuate F2-induced U-87 cell death), required new protein synthesis, and seemed to be regulated by MAPKs. Moreover, we demonstrated the involvement of some Gi/Go protein in F2-triggered calcium mobilization in glioblastoma cells. Since, in a variety of experimental systems, a perturbation of intracellular Ca2+ homeostasis due to increased Ca2+ influx has been found to be an early event in the development of cell injury (CitationOrrenius et al., 1992), it is speculated that Ca2+ influx might be involved in F2-induced glioblastoma death.
The MAPK superfamilies are serine/threonine protein kinases, which play pivotal roles in a broad range of biological functions in many cell types. The ERK pathway is intricately involved in regulating cell growth and differentiation, whereas activation of p38 MAPK is crucial for cellular apoptosis in response to a variety of stress stimuli, such as inflammatory cytokine stress. Interestingly, MAPKs have recently been found to be modulated in apoptotic as well as paraptotic cell signaling. Thus, ERK1/2, typically associated with cell survival, may promote cell death as well (CitationHarper & Wilkie, 2003). For example, some groups have demonstrated that the activation of ERK1/2 is necessary for neuronal cell death in different paradigms, such as cell death induced by hemin (CitationRegan et al., 2001), genistein (CitationLinford et al., 2001), and 6-hydroxydopamine (CitationKulich & Chu, 2001). In support of this, ERK1/2 and p38 pathways were found to play active roles in the paraptotic processes in this system. Thus, various extracellular signals might induce the MAPK pathways to elicit distinct cellular processes such as apoptosis, paraptosis, and proliferation (CitationSperandio et al., 2004), and the final outcome of MAPK activation determined by parallel interactions with several other signaling pathways is very difficult to predict (CitationSubramaniam et al., 2004).
More recently, different PCD pathways have been described, and the form of non-apoptotic programmed cell death paraptosis has been drawing more attention. Paraptosis has been reported in Apaf-1 null mouse embryonic fibroblasts. In addition, neurodegenerative diseases such as Huntington’s disease and amyotrophic lateral sclerosis demonstrate neuronal cell death that does not fulfill the criteria for apoptosis but instead resembles a recently described form of PCD dubbed paraptosis (CitationDal Canto & Gurney, 1994; CitationSperandio et al., 2000; CitationTurmaine et al., 2000). Despite the enormous importance of the discovery of apoptosis as a cell death program indispensable for embryogenesis and protection against unbridled cell growth, the paraptosis pathway is an important safeguard mechanism to protect the organism against unwanted and potential harmful cells when apoptosis routes fail, but can also be triggered in response to cytotoxic agents or other death stimuli (CitationBroker et al., 2005). Yet the precise biochemical mechanism for paraptosis and the basic components of the death machinery are currently unknown (CitationSperandio et al., 2000). Understanding the mechanisms for paraptosis would have potentially important implications for the understanding of evolutionary aspects of cell death programs, developmental cell death, neurodegeneration, and cancer therapeutics, and for the design of novel therapeutic agents for diseases featuring these alternative forms of cell death (CitationBroker et al., 2005).
Conclusions
Taken together, our observations suggest that F2 induces a paraptosis-like programmed cell death in human glioblastoma U-87 cells. Although further work will be necessary to clarify its specific biochemical mechanisms, the current report complements previous studies in the characterization of F2-induced U-87 cell death, enhances our understanding of the action mechanism of F2 on glioma, and helps in the development of novel antitumor therapeutics.
Declaration of interest
This research is partially supported by the Project of Key Laboratory for New Drug Screening of Liaoning Province, and by a national project for New Drug Discovery and Development.
References
- Asare N, Landvik NE, Lagadic-Gossmann D, Rissel M, Tekpli X, Ask K, Lag M, Holme JA (2008): 1-Nitropyrene (1-NP) induces apoptosis and apparently a non-apoptotic programmed cell death (paraptosis) in Hepa1c1c7 cells. Toxicol Appl Pharmacol 230: 175–186.
- Broker LE, Kruyt FA, Giaccone G (2005): Cell death independent of caspases: A review. Clin Cancer Res 11: 3155–3162.
- Bursch W, Hochegger K, Torok L, Marian B, Ellinger A, Hermann RS (2000): Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. J Cell Sci 113: 1189–1198.
- Castro-Obregon S, Del Rio G, Chen SF, Swanson RA, Frankowski H, Rao RV, Stoka V, Vesce S, Nicholls DG, Bredesen DE (2002): A ligand-receptor pair that triggers a non-apoptotic form of programmed cell death. Cell Death Differ 9: 807–817.
- Chen Y, Douglass T, Jeffes EW, Xu Q, Williams CC, Arpajirakul N, Delgado C, Kleinman M, Sanchez R, Dan Q, Kim RC, Wepsic HT, Jadus MR (2002). Living T9 glioma cells expressing membrane macrophage colony-stimulating factor produce immediate tumor destruction by polymorphonuclear leukocytes and macrophages via a “paraptosis”-induced pathway that promotes systemic immunity against intracranial T9 gliomas. Blood 100: 1373–1380.
- Clarke PG (1990): Developmental cell death: Morphological diversity and multiple mechanisms. Anat Embryol (Berl) 181: 195–213.
- Dal Canto MC, Gurney ME (1994): Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am J Pathol 145: 1271–1279.
- Gardner R, Cronin S, Leader B, Rine J, Hampton R (1998): Sequence determinants for regulated degradation of yeast 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell 9: 2611–2626.
- Guo L, Wang LH, Sun B, Yang JY, Zhao YQ, Dong YX, Spranger MI, Wu CF (2007): Direct in vivo evidence of protective effects of grape seed procyanidin fractions and other antioxidants against ethanol-induced oxidative DNA damage in mouse brain cells. J Agric Food Chem 55: 5881–5891.
- Gupta PD, Pushkala K (1999): Importance of the role of calcium in programmed cell death: A review. Cytobios 99: 83–95.
- Hallett MB, Hodges R, Cadman M, Blanchfield H, Dewitt S, Pettit EJ, Laffafian I, Davies EV (1999): Techniques for measuring and manipulating free Ca2+ in the cytosol and organelles of neutrophils. J Immunol Methods 232: 77–88.
- Harper SJ, Wilkie N (2003): MAPKs: New targets for neurodegeneration. Expert Opin Ther Targets 7: 187–200.
- Hoa NT, Zhang JG, Delgado CL, Myers MP, Callahan LL, Vandeusen G, Schiltz PM, Wepsic HT, Jadus MR (2007): Human monocytes kill M-CSF-expressing glioma cells by BK channel activation. Lab Invest 87: 115–129.
- Kulich SM, Chu CT (2001): Sustained extracellular signal-regulated kinase activation by 6-hydroxydopamine: Implications for Parkinson’s disease. J Neurochem 77: 1058–1066.
- Leist M, Jaattela M (2001): Four deaths and a funeral: From caspases to alternative mechanisms. Nat Rev Mol Cell Biol 2: 589–598.
- Linford NJ, Yang Y, Cook DG, Dorsa DM (2001): Neuronal apoptosis resulting from high doses of the isoflavone genistein: Role for calcium and p42/44 mitogen-activated protein kinase. J Pharmacol Exp Ther 299: 67–75.
- Orrenius S, McCabe MJ Jr, Nicotera P (1992): Ca2+-dependent mechanisms of cytotoxicity and programmed cell death. Toxicol Lett 64–65(Spec No): 357–364.
- Regan RF, Wang Y, Ma X, Chong A, Guo Y (2001): Activation of extracellular signal-regulated kinases potentiates hemin toxicity in astrocyte cultures. J Neurochem 79: 545–555.
- Sperandio S, de Belle I, Bredesen DE (2000): An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci USA 97: 14376–14381.
- Sperandio S, Poksay K, de Belle I, Lafuente MJ, Liu B, Nasir J, Bredesen DE (2004): Paraptosis: Mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Differ 11: 1066–1075.
- Spranger I, Sun B, Mateus AM, Freitas VD, Ricardo-da-Silva JM (2008): Chemical characterization and antioxidant activities of oligomeric and polymeric procyanidin fractions from grape seeds. Food Chem 108: 519–532.
- Subramaniam S, Zirrgiebel U, von Bohlen Und Halbach O, Strelau J, Laliberte C, Kaplan DR, Unsicker K (2004): ERK activation promotes neuronal degeneration predominantly through plasma membrane damage and independently of caspase-3. J Cell Biol 165: 357–369.
- Sun B, Belchior GP, Ricardo-da-Silva JM, Spranger MI (1999): Isolation and purification of dimeric and trimeric procyanidins from grape seeds. J Chromatogr A 841: 115–121.
- Sun B, Leandro C, Ricardo da Silva JM, Spranger I (1998): Separation of grape and wine proanthocyanidins according to their degree of polymerization. J Agric Food Chem 46: 1390–1396.
- Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW (2000): Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington’s disease. Proc Natl Acad Sci USA 97: 8093–8097.
- Valamanesh F, Torriglia A, Savoldelli M, Gandolphe C, Jeanny JC, BenEzra D, Behar-Cohen F (2007): Glucocorticoids induce retinal toxicity through mechanisms mainly associated with paraptosis. Mol Vis 13: 1746–1757.
- Wang Y, Li X, Wang L, Ding P, Zhang Y, Han W, Ma D (2004): An alternative form of paraptosis-like cell death, triggered by TAJ/TROY and enhanced by PDCD5 overexpression. J Cell Sci 117: 1525–1532.
- Wyllie AH, Kerr JF, Currie AR (1980): Cell death: The significance of apoptosis. Int Rev Cytol 68: 251–306.
- Ye X, Krohn RL, Liu W, Joshi SS, Kuszynski CA, McGinn TR, Bagchi M, Preuss HG, Stohs SJ, Bagchi D (1999): The cytotoxic effects of a novel IH636 grape seed proanthocyanidin extract on cultured human cancer cells. Mol Cell Biochem 196: 99–108.
- Zhang FJ, Yang JY, Mou YH, Sun BS, Ping YF, Wang JM, Bian XW, Wu CF (2009): Inhibition of U-87 human glioblastoma cell proliferation and formyl peptide receptor function by oligomer procyanidins (F2) isolated from grape seeds. Chem Biol Interact 179: 419–429.