Abstract
Context: Oleanolic acid (OA), a triterpenoid compound, exists in many plants. It has numerous bioactivities and has been used to treat hepatitis in China. However, few studies have reported its effect on the central nervous system, especially in ischemic stroke.
Objective: To explore the protective effects of OA on cerebral ischemic injury for the first time.
Materials and methods: Survival time was tested in mice injured by bilateral common carotid artery ligation (BCCAL). Neurological function, infarct area, cerebral edema, superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), and malondialdehyde (MDA) were estimated in rats operated by middle cerebral artery occlusion (MCAO). Cell survival, lactate dehydrogenase (LDH), SOD, reduced glutathione (GSH), MDA, mitochondrial membrane potential (MMP) and succinic dehydrogenase (SDH) were detected in H2O2-injured PC12 cells.
Results: Pre-administration with OA significantly prolonged survival time in mice at 50 and 25 mg/kg, alleviated neurological function, infarct area and cerebral edema, increased SOD and GSH-Px activities and decreased MDA level in rats at 25 and 12.5 mg/kg. Pre-treatment with OA at 10 and 1 μM remarkably improved cell survival, enhanced SOD activity and GSH content, reduced LDH and MDA levels and reversed the lowering of MMP and SDH activity.
Discussion and conclusion: These results demonstrate that oleanolic acid effectively alleviates cerebral ischemic damage in vivo and oxidative injury in vitro, which may be in part due to the modulation of endogenous antioxidants and the improvement of mitochondrial function. Oleanolic acid may be a potential medicine for attenuating ischemic stroke.
Introduction
Ischemic stroke is a common disorder of the nervous system. It results from transient or permanent reduction in cerebral blood flow, which is caused by the occlusion of the cerebral artery either by an embolus or by local thrombosis (CitationDirnagl et al., 1999). Many pathogeneses are involved in ischemic brain injury, such as energy failure, oxidative stress, excitotoxicity, inflammation and apoptosis and so on (CitationDirnagl et al., 1999; CitationLipton, 1999). Among these mechanisms, oxidative stress induces brain injury by damaging the membrane phospholipids and nucleotides directly and acting as a mediator in other cascades indirectly (CitationEmerit et al., 2004; CitationMargaill et al., 2005). It plays a pivotal and linked role for neuronal death. Therefore, anti-oxidation treatment could be looked upon as a strategy used for stroke therapy. In fact, several antioxidants have been developed in experiment, and some of these have been tested in relevant clinical studies (CitationSinha et al., 2002; CitationLees et al., 2003; CitationShichinohe et al., 2004; CitationToyoda et al., 2004).
Oleanolic acid (3β-hydroxy-olea-12-en-28-oic acid), is a triterpenoid compound which exists widely in many plants in the form of free acid or triterpenoid saponin aglycone (CitationLiu, 1995). It has numerous biological properties with therapeutic potential and has been used to treat human acute and chronic hepatitis in China (CitationLiu, 1995, Citation2005). However, few studies have reported its role in the nervous system. In particular, there is no information about its effect on ischemic stroke. The present study is the first to investigate the therapeutic possibility of OA in this field.
In several previous studies OA was identified as a strong protector against adriamycin-induced lipid peroxidation in liver and heart microsomes (CitationBalanehru & Nagarajan, 1992). Furthermore, it was found to protect against CCl4-induced hepatotoxicity in rats by enhancing the hepatic antioxidant defense system (CitationKim et al., 2005). These studies revealed that OA was able to fight against chemically induced oxidative injuries in several periphery organs. Therefore, it implies that oleanolic acid might be a candidate medicine for stroke treatment if it can protect nerve cells from oxidative damage in ischemic brain injury.
Based on the above inferences, we planned to establish a cerebral ischemia model by BCCAL in mice (CitationWeng et al., 1999) and by MCAO in rats (CitationLonga et al., 1989), and a duplicate oxidative stress model by H2O2 in differentiated pheochromocytoma (PC12) cells (CitationJi & Gao, 2008), which retain the features of dopaminergic neurons and have been widely used for neuroscience studies in vitro (CitationJang & Surh, 2001). In addition, the neuroprotective effect and antioxidative ability of OA were investigated.
Methods
Regents and cell line
Oleanolic acid (powder, purity > 98%) was purchased from Zelang Medical Technology (Nanjing, China). The substance was dissolved in dimethyl sulfoxide (DMSO) as stock solution and stored at 4°C before using for in vitro tests. It was freshly diluted with phosphate-buffered saline (PBS) before the experiment. The final concentration of DMSO in cultured medium was less than 0.1% (V/V), which caused no measurable effect on cell growth. Also, this substance was dissolved in a vehicle (0.5% CMC-Na solution with 2% Tween 80) for in vivo test. Dulbecco’s modified Eagle medium (DMEM) and trypsin were purchased from Gibco BLR (Grand Island, NY, USA). Fetal calf serum was the product of PAA (Pasching, Austria). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) was purchased from Ameresco (Solon, OH, USA). Rhodamine 123 (Rh123) was purchased from Sigma-Aldrich (St. Louis, MO, USA). 2,3,5-Triphenyl tetrazolium chloride (TTC) was obtained from Jingxi Chemical Technology (Shanghai, China). The diagnostic kits used for LDH, protein content, SOD, GSH-Px, GSH, MDA and SDH assay were bought from Jiancheng Bioengineering Institute (Nanjing, China). PC12 cell line was obtained from the Chinese Academy of Sciences (Shanghai). All other chemicals were of analytic grade.
Animals and administration
Thirty male ICR mice (26 ∼ 30 g) and 56 male SD rats (260 ∼ 300 g) were obtained from the Medical Animal Center of the China Pharmaceutical University. All the animals were kept in a controlled environment at 22° ± 2°C with a 12 h light/dark cycle and free access to food and water, and acclimatized for three days prior to use. All the procedures followed principles in the Declaration of Helsinki and were carried out in compliance with the Regulations of Experimental Animal Administrations issued by the State Committee of Science and Technology of the People’s Republic of China.
According to the different treatments, the mice were divided randomly into three groups, ten in each group, including: control group (BCCAL + vehicle, 20 ml/kg), high dose of OA group (BCCAL + OA, 50 mg/kg) and low dose of OA group (BCCAL + OA, 25 mg/kg). The rats were divided into four groups randomly, 14 in each group, including: sham-operated group (without MCAO + vehicle, 10 mL/kg), model group (MCAO + vehicle, 10 mL/kg), high dose of OA group (MCAO + OA, 25 mg/kg) and low dose of OA group (MCAO + OA, 12.5 mg/kg). All the animals were dosed with OA or vehicle once daily for four days by intraperitoneal injection (i.p.) before the operation.
Operation of bilateral common carotid artery ligation and survival time test
The mice were anesthetized with ethyl ether 30 min after the last administration. The model of acute cerebral ischemia was established by the operation of BCCAL with vagus nerves (CitationWeng et al., 1999). Briefly, bilateral common carotid arteries (BCCA) with vagus nerves were separated free from the surrounding fascia. They were ligated with 3-0 silk sutures tightly. After which, survival time (unit: s) was observed with a stop watch.
Operation of middle cerebral artery occlusion
The rats were anesthetized with 3% chloral hydrate (300 mg/kg, i.p.) 30 min after the last administration. The model of focal cerebral ischemia-reperfusion (FCI/R) was duplicated by the operation of MCAO, which was performed according to the method described previously (CitationLonga et al., 1989). Briefly, the right common carotid artery (CCA), external carotid artery (ECA), occipital artery (OCA), superior thyroid artery (STA) and internal carotid artery (ICA) were exposed and dissected free from surrounding nerves and fascia. OCA, STA and the distal end of ECA were ligated with 3-0 silk sutures and cut off. The mobilized end of ECA was stretched to a line with ICA, and a 40 mm length of 3-0-nylon filament was inserted through the proximal ECA into ICA until the middle cerebral artery (MCA). The filament was inserted 18 ± 0.5 mm from the bifurcation of CCA and slight resistance can be felt. After 2 h occlusion of MCA, the filament was removed to ensure the 24 h reperfusion. If the rats showed signs of recovery during the procedure, they would be anesthetized with 3% chloral hydrate (75 mg/kg, i.p.) immediately. The sham-operated rats were operated identically without occlusion of MCA. Penicillin was applied in the incision to prevent infection before suture. Body temperature was maintained at 37° ± 0.5°C throughout the procedure.
Estimation of neurological function, infarct area and cerebral edema
Six rats from each group were used for these estimations. Deficit score was noted after 24 h reperfusion, by an investigator who was blind to the treatment regimen, according to the standard described previously by CitationLonga et al. (1989): 0, no neurological deficits; 1, failure to extend left paw fully; 2, circling to left; 3, falling to left; 4, show lower levels of consciousness and cannot walk spontaneously.
After the neurological function test, these rats were decapitated. Their brains were removed quickly, washed with cold PBS and dried with filter papers. After weighing the brains, they were rapidly frozen and cut into 5 slices of about 2 mm thickness and stained with 1% TTC in PBS at 37°C for 10 min. After photographing, the pallid areas were separated, dried with filter papers and weighed. The infarct area was expressed as a percentage and calculated by the formula:
After measuring the infarct area, the brain slices were dried in an oven at 110°C to obtain constant dry weight. Cerebral edema was estimated by water content, which was expressed as a percentage and calculated as following:
Measurement of SOD, GSH-Px and MDA in ischemic hippocampus
The other eight rats of each group were decapitated and used for these measurements. The hippocampus of the ischemic hemisphere was removed quickly and homogenized in 0.9% NaCl on an ice board. The homogenate was centrifugated at 3000 rpm for 10 min at 4°C. The supernatant was collected for the measurement of SOD, GSH-Px and MDA according to the corresponding kits’ instructions.
Cell culture and treatment
Nerve growth factor (NGF)-differentiated PC12 cells were cultured in DMEM supplemented with 10% of fetal calf serum and 100 units/mL penicillin and 100 units/mL streptomycin in a humidified cell incubator at 37°C in 5% CO2 and 95% air. All experiments were carried out 24 h after the cells were seeded onto plates or flasks at 1 × 105 cells/mL. H2O2 was freshly prepared prior to each experiment. Cells were pre-incubated with OA in different final concentrations (10, 1, 0.1 μM) for 1 h before exposure to H2O2 in a final concentration of 300 μM for 2 h. The control groups were given equal volume of PBS without OA and H2O2. And then assays for cell survival, activities of LDH, SOD and SDH, GSH and MDA contents and MMP were performed.
Determination of cell survival and analysis of LDH
Cells were seeded in 96-well plates for determination of cell survival by the reduction of MTT or in 24-well plates for analysis of LDH.
After the injury was completed, MTT was added to each well in a final concentration of 0.5 mg/mL for further incubation. After 4 h the medium was removed and the formazan crystals were dissolved in DMSO (150 μL/well) with shaking for 15 min to ensure complete solubilization. Finally, the absorbance at 570 nm (A570) was measured with a microplate reader. The inhibition rate (IR) of cell damage was calculated by the formula:
At the end of injury the medium was collected for determining LDH by using an assay kit according to the instructions. The inhibition rate (IR) of cell damage in LDH was calculated as following:
Analysis of SOD, GSH and MDA in cells
Cells were seeded in 24-well plates. All treatments were the same as above. After exposure to H2O2, the medium was collected for determination of MDA. Cells were washed twice with ice-cold PBS, and then, they were lysed in a buffer (0.2% Triton X-100 in PBS, pH 7.4) on an ice board. After that the buffer was homogenized, collected and centrifugated at 1000 rpm for 5 min at 4°C. The supernatant was collected for SOD, GSH and protein content assay. All the detections were performed with spectrophotometry according to the corresponding kits’ instructions.
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential was determined by Rh123, which is a cell-permeable cationic fluorescent dye. Cells were prepared as described above except that they were seeded in flasks. At the end of H2O2-induced injury, cells were collected and resuspended in PBS at 1 × 106 cells/mL, and then incubated with 1 μM Rh123 for 30 min in darkness. Cells were washed twice with PBS after the incubation. The fluorescence intensity of Rh123 was measured with flow cytometry, the excitation or emission filter was set at 480 nm or 530 nm, respectively.
Analysis of SDH
Cells were seeded in 6-well plates and prepared as described above. After exposure to H2O2, the supernatant, which was acquired as described above, was collected for SDH assay. The procedure was performed as per the diagnostic kits’ instruction.
Statistical analysis
When necessary, data were expressed as mean ± SD, Grubbs’ test was used for assessing outliers and homogeneity of variance was analyzed by Bartlett’s test, and then Student’s t-test or Welch’s t-test was used for single comparison according to the results of Bartlett’s test. Differences were considered significant at P <0.05.
Results
Effect of oleanolic acid on survival time in acute cerebral ischemic mice
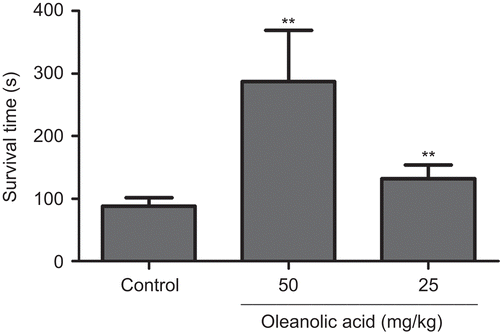
The model of acute cerebral ischemia-induced ischemic hypoxia in mouse was used to assess the anti-hypoxia effect of oleanolic acid. Two outliers in each group were excluded according to Grubbs’ test. As shown in , both high and low doses of OA (50 and 25 mg/kg) significantly (P <0.01) prolonged the survival time of ischemic hypoxia mice to 286.78 ± 82.53 and 131.90 ± 21.69 s, respectively, in comparison with the control group (88.25 ± 12.99 s). It shows that OA has the property of anti-ischemic hypoxia in ICR mice.
Figure 1. Survival time in acute cerebral ischemia mice (n = 8). **P <0.01 versus control.
Effects of oleanolic acid on neurological function, infarct area and cerebral edema in focal cerebral ischemia-reperfusion rats
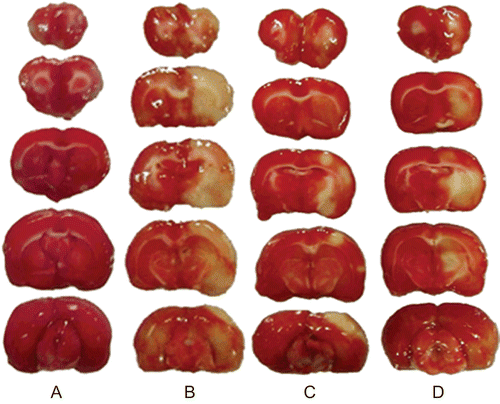
The photographs of TTC staining are shown in . Pre-administration with OA appears to reduce the pallid area compared with the model group. The other results are shown in . Compared with the sham-operated group, the neurological deficit score, infarct area and water content of model group were increased significantly (P <0.01). High dose of OA (25 mg/kg) can reverse all the changes obviously (P <0.01 or P <0.05). And low dose of OA (12.5 mg/kg) can decrease the infarct area and water content notably (P <0.05). The data suggest that OA can protect SD rats from injury of focal cerebral ischemia-reperfusion effectively.
Table 1. Effects of oleanolic acid on neurological function, infarct area and cerebral edema in focal cerebral ischemia-reperfusion rats (X ± SD, n = 6).
Figure 2. Brain slices were stained by 1% TTC solution. (A) Sham-operated group. (B) Model group. (C to D) Rats were dosed with oleanolic acid at 25 or 12.5 mg/kg for four days before MCAO respectively.
Antioxidant activity of oleanolic acid in focal cerebral ischemia-reperfusion rats
Measurements of SOD, GSH-Px and MDA were used to evaluate the antioxidant activity of OA in this rat model. As shown in , occlusion and reperfusion of MCA caused a decrease in the activities of SOD and GSH-Px and an increase in MDA level significantly (P <0.01). However, pre-administration with OA (25 and 12.5 mg/ kg) attenuated the changes of SOD, GSH-Px and MDA significantly (P <0.01 or P <0.05).
Table 2. Antioxidant activity of oleanolic acid in focal cerebral ischemia-reperfusion rats (X ± SD, n = 8).
Oleanolic acid protected PC12 cells against oxidative stress-induced cytotoxicity and LDH leakage
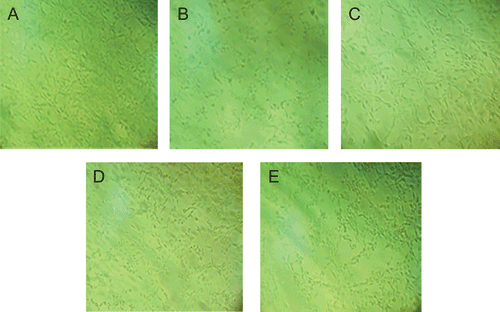
Cell survival was evaluated by the reduction of MTT. The release of LDH, which is another indicator of cell toxicity, was determined for further investigation. The photographs of cell morphology are shown in . Results are shown in . Compared with the control group a significant (P <0.01) reduction in A570 or an increase in LDH leakage was observed in the model group. After pre-incubation with OA at 10, 1 and 0.1 μM, the reduced A570 or increased LDH release was attenuated obviously (P <0.01 or P <0.05). The results show that OA may protect PC12 cells against H2O2-induced injury effectively.
Table 3. Effect of oleanolic acid on oxidative stress induced cytotoxicity and LDH leakage in PC12 cells (X ± SD).
Figure 3. PC12 cells were observed by inverted microscope (100×). Cells were treated with H2O2 in the absence or in the presence of oleanolic acid except for the control group. (A) Control conditions. (B) After exposure to 300 μM H2O2 for 2 h. (C to E) Cells were pretreated with oleanolic acid for 1 h at 10, 1, 0.1 μM before exposure to H2O2.
Antioxidant activity of oleanolic acid in H2O2-injured cells
Determination of SOD, GSH and MDA was used to evaluate the antioxidant activity of OA in this model. As shown in , treatment of PC12 cells with 300 μM H2O2 for 2 h caused a decrease in SOD activity and GSH content and an increase in MDA level significantly (P <0.01 or P <0.05). However, pre-treatment with 10 and 1 μM OA ameliorated the changes of SOD, GSH and MDA remarkably (P <0.05).
Table 4. Effect of oleanolic acid on antioxidative indicators in H2O2 injured PC12 cells. (X ± SD, n = 3).
Effect of oleanolic acid on MMP of cells injured by H2O2
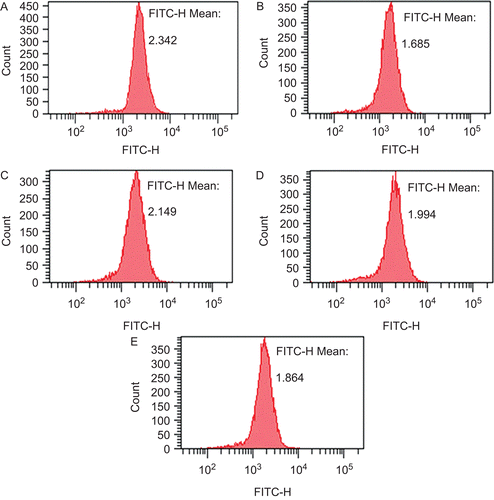
Measurement of MMP was used to assess the structural damage in mitochondrial membrane. MMP was determined by the fluorescence intensity of Rh123. The results of fluorescence intensity are shown in . After incubation with 300 μM H2O2 for 2 h, MMP was decreased remarkably. However, pre-treatment with OA for 1 h reversed the lowering of MMP obviously.
Figure 4. Effect of oleanolic acid on MMP of cells injured by H2O2. Cells were treated with H2O2 in the absence or in the presence of oleanolic acid except for the control group. (A) Control conditions. (B) After exposure to 300 μM H2O2 for 2 h. (C to E) Cells were pretreated with oleanolic acid for 1 h at 10, 1, 0.1 μM before exposure to H2O2.
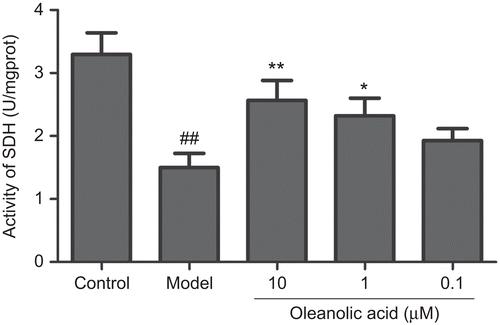
Effects of oleanolic acid on activity of SDH in H2O2-injured cells
SDH, which is localized in mitochondrial inner membrane, is a key enzyme that participates in the tricarboxylic acid cycle and electron transport chain. Analysis of SDH was used to assess the function of mitochondrion. From we can observe that incubation with 300 μM H2O2 for 2 h caused a significant (P <0.01) decrease in SDH activity (unit: U/mgprot) from 3.3 ± 0.34 to 1.5 ± 0.22. However, pre-treatment with OA at 10, 1 and 0.1 μM remarkably increased SDH activity to 2.57 ± 0.31 (P <0.01), 2.32 ± 0.28 (P <0.05) and 1.93 ± 0.19 respectively.
Figure 5. Effect of oleanolic acid on activity of SDH in H2O2 injured PC12 cells (n = 3). ##P <0.01 versus control; **P <0.01, *P <0.05 versus model.
Discussion
As described in the introduction, anti-oxidation treatment has been developed in experiments and tested in clinical studies. Oleanolic acid was proved to be very effective in chemically induced oxidative injuries in heart and liver. We propose that oleanolic acid may have potential against cerebral ischemia injury if it can protect nerve cells from oxidative damage in this pathological condition.
Brain tissue utilizes approximately 20% of the total oxygen supply of the body. Its energy substrate supply primarily depends on glucose. In normal brain, cellular energy is produced almost exclusively by oxidative phosphorylation and conserved in the form of ATP, which is necessary for cell existence. Impairment of cerebral blood flow restricts the delivery of oxygen and glucose, resulting in a decrease in ATP formation and subsequent energy failure, which form the early events leading to further damage during cerebral ischemia injury (CitationMartin et al., 1994; CitationMargaill et al., 2005). Thus, the property of OA on anti-ischemic hypoxia was tested for initial drug screening. Permanent ligation of BCCA with vagus nerves in mice led to serious obstruction of cerebral blood supply, and subsequently caused death. The result showed OA (50 and 25 mg/kg) significantly prolonged the survival time of brain ischemic mice compared with the control group. This led us to continue further our studies with OA.
Beginning with the energy failure, the ionic gradients are disrupted and characterized by influx of Na+ and Ca2+, efflux of K+ and cellular depolarization. Voltage-dependent Ca2+ channel becomes activated and excitatory amino acids, such as glutamate, are released into the extracellular space. Glutamate contributes to a further influx of Ca2+, Na+ and Cl− through different receptors. Water follows passively causing the cell to swell and cerebral edema. The increased Ca2+ causes mitochondrial Ca2+ overload leading to the leakage of mitochondrial membrane, interruption of electron transport chain, cessation of already compromised ATP production, and burst of reactive oxygen species (ROS) (CitationKoistinaho & Koistinaho, 2005). ROS consist of oxygen free radicals and other associated elements, such as superoxide free radicals, hydrogen peroxide, etc. (CitationChong et al., 2005). This contributes to membrane lipid peroxidation, damage to DNA and protein and production of inflammatory mediators (CitationKoistinaho & Koistinaho, 2005). All these injuries lead to neuronal death in the infarct area and result in neurological function deficits. Oxidative stress plays a pivotal and linked role in these pathologies. Endogenous antioxidants play an integral role in removing excess ROS (CitationCrack & Taylor, 2005). SOD is able to transform superoxide radicals into H2O2 and the formed H2O2 is subsequently scavenged by GSH and GSH-Px. There is mounting evidence through the generation of transgenic and knockout mutant mice which suggests that these antioxidants play a key protective role in stroke (CitationCrack & Taylor, 2005).
In our study, OA (25 and 12.5 mg/kg) significantly alleviated neurological deficit score, infarct area and cerebral water content in SD rat, compared with the model group. Furthermore, OA significantly enhanced the activities of SOD and GSH-Px and decreased MDA level in ischemic hippocampus, which is a vulnerable structure to ischemic injury (CitationHomi et al., 2002). To further examine the anti-oxidative ability of OA, an in vitro model of oxidative stress in PC12 cell was duplicated by H2O2. The results showed that pre-treatment with OA at 10 and 1 μM protected PC12 cells from H2O2-induced injury by improving cell survival, enhancing SOD activity and GSH content and reducing LDH and MDA level. It means OA possesses good neuroprotective effect and antioxidant action in SD rats injured by focal cerebral ischemia-reperfusion and in PC12 cells injured by H2O2.
During ischemic brain injury, energy failure, Ca2+ overload and some other upstream events destroy the mitochondria, impair the respiratory chain and lead to the generation of massive ROS, which overrides the clearance ability of endogenous antioxidants, and thus contributing to oxidative stress. ROS, mainly generated in mitochondria (CitationEmerit et al., 2004), can interact with unsaturated fatty acids in mitochondria membrane to formulate saturated fatty acids and fatty acid free radicals which cause the reduction of membrane fluidity, the enhancement of membrane permeability and the lowering of MMP (CitationSomayajulu et al., 2005). In addition, they can also damage some important respiratory enzymes, inhibit the electron transport chain (ETC) and tricarboxylic acid cycle (TCA), resulting in the decrease of ATP (CitationGibson & Huang, 2004). Furthermore, ROS lead to the loss or breakage of mtDNA and influence transcription subsequently. All these injuries can block oxidation phosphorylation, which leads to more ROS production and forms a vicious cycle (CitationBelogrudov & Hatefi, 1994). In addition, mitochondrial permeability transition pore activated by ROS causes the release of cytochrome c and some other apoptotic factors, inducing multiple apoptotic pathways (CitationEmerit et al., 2004). It is clear that mitochondria, important for energy conversion in eukaryocytes, are not only major contributors to oxidative stress, but also sensitive sufferers to the injuries. Therefore, the protection of mitochondria is very important.
In the present study, staining brain sections with TTC, which is colored red by electron transport in active mitochondria, identified a region in which mitochondrial function is severely compromised (CitationLipton, 1999). The changes of MMP and SDH in cells were used to assess the structural and functional damage of mitochondria. As shown in the results, OA reduced the pallid area in rat brains apparently, and reversed the decreased MMP and SDH in PC12 cells significantly. It suggests that OA may improve the mitochondrial function destroyed by FCI/R in SD rats and by H2O2 in PC12 cells.
Taken together, these results provide a new insight into the neuroprotective action of oleanolic acid. However, the mechanism underlying the protective effect of OA on cerebral ischemia injury has not resolved yet. Recent study has revealed that oleanolic acid facilitated nuclear factor-erythroid 2-related factor 2 (Nrf2) nuclear accumulation, causing induction of Nrf2-dependent genes, which contributes to protection from acetaminophen hepatotoxicity in mice (CitationReisman et al., 2009). Nrf2 is a nuclear transcription factor. When exposed to ROS, activated Nrf2 translocates to the nucleus, where it binds to antioxidant response elements (ARE) in the promoter region of genes encoding antioxidant enzymes, thereby inducing the production of enzymes such as SOD, GSH-Px, catalase and so on (CitationVries et al., 2008). Several reports have confirmed the beneficial effects of dietary tert-butylhydroquinone (tBHQ) and sulforaphane, both potent inducers of the Nrf2-ARE pathway, in animal models of cerebral ischemia (CitationShih et al., 2005; CitationZhao et al., 2006). It implies that there might be an indefinite relationship between the Nrf2-ARE pathway and the protective effect of OA on cerebral ischemia injury. Related studies will be reported in the future.
Conclusions
In summary, our study demonstrates for the first time that oleanolic acid alleviates effectively cerebral ischemic damage in mice and rats, and protects PC12 cells from H2O2-induced injury. The mechanism may be, at least in part, attributable to the modulation of endogenous antioxidants and the improvement of mitochondrial function. These findings indicate that oleanolic acid may be beneficial for attenuating cerebral injury in ischemic stroke.
Declaration of interest
The present study was supported by the National Natural Science Foundation of China (NO. Y071013-1). All the authors declare that they have no financial and personal relationships with other people or organizations that could inappropriately influence their work. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Balanehru S, Nagarajan B. (1992). Intervention of adriamycin induced free radical damage. Biochem Int, 28, 735–744.
- Belogrudov G, Hatefi Y. (1994). Catalytic sector of complex I (NADH: ubiquinone oxidoreductase): Subunit stoichiometry and substrate-induced conformation changes. Biochemistry, 33, 4571–4596.
- Chong ZZ, Li FQ, Maiese K. (2005). Oxidative stress in the brain: Novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol, 75, 207–246.
- Crack PJ, Taylor JM. (2005). Reactive oxygen species and the modulation of stroke. Free Radic Biol Med, 38, 1433–1444.
- Dirnagl U, Iadecola C, Moskowitz MA. (1999). Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci, 22, 391–397.
- Emerit J, Edeas M, Bricaire F. (2004). Neurodegenerative diseases and oxidative stress. Biomed & Pharmacother, 58, 39–46.
- Gibson GE, Huang HM. (2004). Mitochondrial enzymes and endoplasmic reticulum calcium stores as targets of oxidative stress in neurodegenerative diseases. J Bioenerg Biomembr, 36, 335–340.
- Homi HM, Freitas JJS, Curi R, Velasco IT, Junior BAS. (2002). Changes in superoxide dismutase and catalase activities of rat brain regions during early global transient ischemia/reperfusion. Neurosci Lett, 333, 37–40.
- Jang JH, Surh YJ. (2001). Protective effects of resveratrol on hydrogen peroxide-induced apoptosis in rat pheochromocytoma (PC12) cells. Mutat Res, 496, 181–190.
- Ji BS, Gao Y. (2008). Protective effect of trihexyphenidyl on hydrogen peroxide-induced oxidative damage in PC12 cells. Neurosci Lett, 437, 50–54.
- Kim NY, Lee MK, Park MJ, Kim SJ, Park HJ, Choi JW, Kim SH, Cho SY, Lee JS. (2005). Momordin Ic and oleanolic acid from Kochiae Fructus reduce carbon tetrachloride-induced hepatotoxicity in rats. J Med Food, 8, 177–183.
- Koistinaho M, Koistinaho J. (2005). Interactions between Alzheimer’s disease and cerebral ischemia – Focus on inflammation. Brain Res Rev, 48, 240–250.
- Lees KR, Barer D, Ford GA, Hacke W, Kostulas V, Sharma AK, Odergren T. (2003). Tolerability of NXY-059 at higher target concentrations in patients with acute stroke. Stroke, 34, 482–487.
- Lipton P. (1999). Ischemic cell death in brain neurons. Physiol Rev, 79, 1431–1568.
- Liu J. (1995). Pharmacology of oleanolic acid and ursolic acid. J Ethnopharmacol, 49, 57–68.
- Liu J. (2005). Oleanolic acid and ursolic acid: Research perspectives. J Ethnopharmacol, 100, 92–94.
- Longa EZ, Weinstein PR, Carlson S, Cummins R. (1989). Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke, 20, 84–91.
- Margaill I, Plotkine M, Lerouet D. (2005). Antioxidant strategies in the treatment of stroke. Free Radic Biol Med, 39, 429–443.
- Martin RL, Lloyd HGE, Cowan AI. (1994). The early events of oxygen and glucose deprivation: Setting the scene for neuronal death? Trends Neurosci, 17, 251–257.
- Reisman SA, Aleksunes LM, Klaassen CD. (2009). Oleanolic acid activates Nrf2 and protects from acetaminophen hepatotoxicity via Nrf2-dependent and Nrf2-independent processes. Biochem Pharmacol, 77, 1273–1282.
- Shichinohe H, Kuroda S, Yasuda H, Ishikawa T, Iwai M, Horiuchi M, Iwasaki Y. (2004). Neuroprotective effects of the free radical scavenger Edaravone (MCI-186) in mice permanent focal brain ischemia. Brain Res, 1029, 200–206.
- Shih AY, Li P, Murphy TH. (2005). A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci, 25, 10321–10335.
- Sinha K, Chaudhary G, Gupta YK. (2002). Protective effect of resveratrol against oxidative stress in middle cerebral artery occlusion model of stroke in rats. Life Sci, 71, 655–665.
- Somayajulu M, McCarthy S, Hung M, Sikorska M, Borowy-Borowski H, Pandey S. (2005). Role of mitochondria in neuronal cell death induced by oxidative stress; Neuroprotection by coenzyme Q10. Neurobiol Dis, 18, 618–627.
- Toyoda K, Fujii K, Kamouchi M, Nakane H, Arihiro S, Okada Y, Ibayashi S, Iida M. (2004). Free radical scavenger, edaravone, in stroke with internal carotid artery occlusion. J Neurol Sci, 221, 11–17.
- Vries HED, Witte M, Hondius D, Rozemuller AJM, Drukarch B, Hoozemans J, Horssen JV. (2008). Nrf2-induced antioxidant protection: A promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic Biol Med, 45, 1375–1383.
- Weng HB, Ma T, You YR, Liu YL. (1999). Effects of daidzein on anoxia and cerebral ischemia in mice. J Shenyang Pharm Univ, 16, 63–64.
- Zhao J, Kobori N, Aronowski J, Dash PK. (2006). Sulforaphane reduces infarct volume following focal cerebral ischemia in rodents. Neurosci Lett, 393, 108–112.