Abstract
Context: The kringle 2 plus serine protease domains (K2S) of human tissue plasminogen activator (tPA) is an efficacious thrombolytic drug, which has been used to treat heart attacks and strokes by breaking up the clots that cause them. It has nine disulfide bridges, which are needed for proper folding and be the bottleneck in improving the production in the Escherichia coli system. So far, few reports have described the production of soluble active K2S from E. coli.
Objective: To achieve high-level expression of active K2S in the E. coli system.
Materials and methods: The DNA fragment coding for K2S was fused with the E. coli disulfide isomerase DsbC. The constructed fusion protein was expressed in E. coli, and then purified with the Ni2+-chelating affinity chromatography. K2S was released by cleavage with Factor Xa protease, and the thrombolytic activity was determined using the fibrin plate assay.
Results: The fusion protein DsbC-K2S was found in the culture supernatant of recombinant E. coli as a soluble form of ~40%. The result of fibrinolysis fibrin plate assay showed that the purified recombinant K2S exhibited significant fibrinolysis activity in vitro.
Discussion and conclusion: These works provided a novel approach for the production of active K2S in E. coli without the requirements of in vitro refolding process, and might establish a significant foundation for the following production of K2S.
Introduction
Thrombosis occurs when a clot inside a blood vessel restricts or blocks blood flow. It can lead to heart attacks or strokes, which are two of the major causes of death in the world. Thrombolytic therapy is one of the most effective, lifesaving treatments for such diseases (CitationAdams, 2003; CitationBaker, 2005).
The most commonly used drug for thrombolytic therapy is tissue plasminogen activator (tPA, Alteplase). tPA is a serine protease sequentially consists of five distinct structural domains: a fibronectin-like finger domain (F), an epidermal growth factor-like domain (E), two disulfide-bonded triple-loop structures commonly called kringles (K1 and K2), and, at the carboxyl terminus, a serine protease domain (S) (CitationPennica et al., 1983; CitationKalyan et al., 1988). Previous studies have indicated that the F domain and the K2 domain are involved in fibrin binding of tPA, and the S domain is responsible for converting plasminogen to plasmin, which is important for the homeostasis of fibrin formation and clot dissolution (Citationvan Zonneveld et al., 1986; CitationRenatus et al., 1997). To improve the clinical efficacy and facilitate the production, many smaller variants of tPA have further been considered (CitationLarsen et al., 1989; CitationRenatus et al., 1997).
The deletion-mutant variant including K2 plus S domain of human tPA (K2S, rPA, Reteplase) is modified to contain amino acids 1–3 and 176–527 of the original protein. K2S is similar to tPA, but has a longer half-life of 13–16 min than tPA (4–8 min), and it also binds fibrin with lower affinity than tPA, improving its ability to penetrate into clots (CitationWooster & Luzier, 1999; CitationQureshi et al., 2002; CitationSimpson et al., 2006). Compared with tPA, K2S is a nonglycosylated protein and more suitable for production in the E. coli system, the most simple and cost-effective recombinant protein expression system nowadays. However, the K2S polypeptide has nine disulfide bridges, which are needed for proper folding and be the bottleneck in improving the production in the E. coli system. To achieve the production of active K2S in E. coli, in this study, the K2S coding sequence was amplified from the tPA gene and cloned into the downstream of the disulfide-bond isomerase (DsbC) gene. The fusion protein was expressed in E. coli with isopropyl-1-thio-β-galactopyranoside (IPTG) induction, and then purified by Ni2+-chelating affinity chromatography. The mature K2S was excised from fusion protein by Factor Xa protease cleavage, and its thrombolytic activity was also investigated.
Materials and methods
Bacterial strains, plasmids, and growth conditions
For cloning purposes, E. coli strains DH5α and plasmid pET40b (+) were used. The E. coli BL21 was used for overproduction of the recombinant protein. The growth media used in this work was L-broth (per liter, 5 g NaCl, 5 g yeast extract, and 10 g tryptone; Oxoid). Kanamycin (50 µg/ml) was used to select for the recombinant bacteria.
Amplification of the K2S gene by PCR
In order to amplify the coding sequence of K2S, a pair of primers, K2S-1 (5′-CGGGGATCCGATCGAAGGTCGTTCTTACCAAGGAAACAGTG-3′) and K2S-2 (5′-GGGCTCGAGATTACGGTCGCATGTTGTCACGAATCCAG-3′), were synthesized. These primers were designed based on the human tPA gene retrieved from the NCBI GenBank. The primer K2S-1 incorporated a BamHI site and Factor Xa protease cleavage sequence, and the primer K2S-2 incorporated a XhoI site and a translational termination codon at their 5′-terminus.The pMD18T-tPA recombinant plasmid constructed in our previously studies was used as the template (CitationGang et al., 2006). The PCR was performed at 94°C for 5 min, then 28 cycles at 94°C for 45 s, at 64°C for 45 s, and at 72°C for 1.5 min; extension was carried out at 72°C for 10 min.
Construction of the DsbC-K2S expression plasmid
The 1.1-kb PCR product digested with BamHI and XhoI was subcloned into pET40b (+) plasmid to generate a fusion protein with DsbC. The recombinant construction was analyzed by restriction-enzyme digestion and sequencing to determine reading frame orientation and confirm sequence fidelity, and the positive recombinant plasmid was named as pET40b (+)-K2S. In addition, between DsbC and K2S coding sequence, there was a His-Tag, which was beneficial for the fusion protein separation by Ni2+-chelating affinity chromatography.
Expression of K2S in shaker flasks
The recombinant plasmid pET40b (+)-K2S was transformed into E. coli strain BL21 for expression. In brief, the transformant was inoculated in 100 ml of Luria-Bertani medium at pH 7.0 in the presence of kanamycin (50 mg/ml) at 37°C. When OD600 absorption reached 0.6–1.0, 0.6 mM IPTG was added to induce the protein synthesis. The bacteria were further cultured by shaking (200 r.p.m.) for 12 h at 30°C. The bacterial cells were harvested by centrifugation at 13,000g for 20 min, resuspended in 25 ml of 20 mM Tris-HCl (pH 7.9), and then lysed by sonication (Ultrasonic Cell Crusher) at 400W for 30 min (4 s working, 6 s free) in an ice-water bath. The content of protein was determined with the method of bicinchoninic acid assay.
Purification of the recombinant fusion protein
The supernatant of the cell lysate resulting from centrifugation was applied to a column packed with 10 ml of Ni2+-chelating resin that had been previously equilibrated with NTA-0 buffer (0.5 M NaCl, 20 mM Tris-HCl, 10% glycerol, pH 7.9). After washing to baseline absorbance with NTA-0 buffer, the column was washed with elution buffer containing 10, 20, 50, 100, 200, 300, and 400 mM imidazole at a flow rate of 2 ml/min. Each peak were collected and subjected to SDS-PAGE, and the relative quantities of fusion product were measured with the Quantity One software.
For cleavage of K2S from DsbC tag, the purified fusion protein was cleaved by Factor Xa protease. As the DsbC still followed with a His-Tag but the mature K2S without, Ni2+-chelating affinity chromatography was performed again to purify the mature K2S from the cleaved protein solution. The mature K2S was obtained by centrifugation.
Thrombolytic activity assay for K2S
The fibrin plates was used to assay the activity of the K2S. Briefly, 0.02 g fibrin was dissolved in 5 ml phosphate-buffered saline (PBS) by mixing softly and heating at 37°C for 10 min, then the thrombin was added. Agarose 0.07 g was dissolved with 7 ml PBS, mixed with the fibrin solution anterior referred, and then poured to flat-panels softly. Subsequently, the stiletto method was used, and the thrombolytic activity was determined by measuring the diameter of the clear circle surrounding the sample spot. Both the DsbC-K2S fusion protein (125 μg) and K2S protein after Factor Xa cutting (50 μg) were detected. In the meantime, tPA (10 U) and urokinase (10 U) were assayed as the positive control, and the expression product of pET40b (+) was also examined as the negative control. The thrombolytic activity corresponding to tPA of the recombinant product was calculated as follows:
In the above formula, D was the diameter of the clear circle surrounding the spot. U was the thrombolytic activity unit of tPA (1,000 U/ml in the present study). V was the volume of the agents.
Results
Construction of DsbC-K2S fusion plasmid
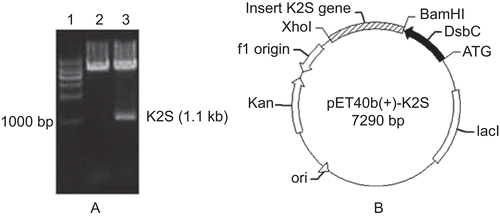
The coding sequence of K2S was amplified from the pMD18T-tPA plasmid and inserted into the pET40b (+) vector. The results of restriction digestion showed that the target fragment had been successfully inserted into the vector, and the following DNA sequencing also validated that the DsbC gene and the K2S coding sequence were combined into one open reading frame (). The map of DsbC-K2S recombinant vector was shown in .
Figure 1. Construction of DsbC-K2S fusion plasmid. (A) Identification of DsbC-K2S fusion plasmid by restriction digestion. Lane 1: 1-kb DNA marker. Lane 2: pET40b plasmid digested with BamHI and XhoI. Lane 3: DsbC-K2S plasmid digested with BamHI and XhoI. (B) Map of the DsbC- K2S fusion plasmid.
Optimization of K2S expression in shaker flasks
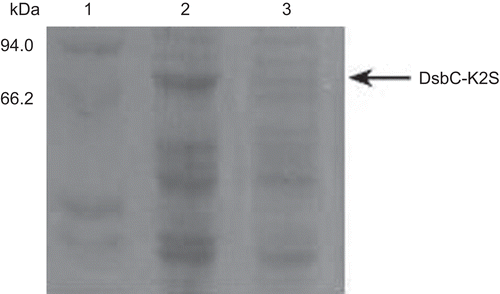
The K2S expression in the recombinant E. coli was induced by IPTG. In order to obtain optimal expression conditions, three factors, including the induction time, the concentration of IPTG and the temperature, were considered. The maximum DsbC-K2S fusion protein production was obtained at the yield of 76.71 mg/l broth when the induction was performed with 0.6 mM IPTG at 25°C for 3 h (). Furthermore, the results of relative quantities measured by the Quantity One software revealed that the DsbC-K2S fusion protein expressed solubility at a high yield reaching about 40% of the total bacteria protein.
Figure 2. The optimal expression conditions on K2S expression. Lane 1: protein marker. Lane 2: the expression product of pET40b (+)-K2S induced by optimal expression conditions. Lane 3: the expression product of pET40b (+) vector.
Purification of the recombinant fusion protein
The recombinant fusion protein sequentially consisted of the following regions from its N-terminus: the DsbC domain, the linker sequence of His-Tag, Factor Xa recognition sites, and K2S domain. As histidine tag could specifically interact with Ni2+, the Ni2+-chelating affinity chromatography was used to purify the target fusion protein. The results of SDS-PAGE showed that the fusion protein DsbC-K2S could be eluted efficaciously from the column with elution buffer containing 10 mM imidazole ( and ).
Figure 3. The elution curve of the Ni2+-chelating affinity chromatography. Fifty microliters of buffer NTA-0 was used to balance the column, then the sample was added. From 0 to 22.5 min was the penetration. After eluted with the elution buffer gradiently. The target protein was obtained at 46–73 min when the imidazole concentration reached at 10 mM.

Figure 4. The SDS-PAGE of the purified DsbC-K2S fusion protein. Lane 1: protein marker; lane 2: the supernatant of the recombinant cell lysate; lane 3: penetration; lane 4: purified DsbC-K2S fusion protein.
Proteolytic cleavage and purification of recombinant K2S
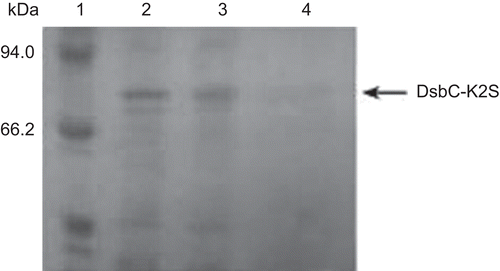
The fusion protein was desalted and subsequently cleaved by Factor Xa in freshly prepared cleavage buffer (pH 8.0) at 23°C for 16 h. The result cleaved solution was applied to SDS-PAGE (). On the SDS-PAGE gel, we observed the target protein band (corresponding to 40 kDa), and another band which was identical to that of DsbC (corresponding to 34 kDa).
Figure 5. SDS-PAGE analysis of the cleaving of the fusion protein. Lane 1: DsbC-K2S fusion protein cleaved by Factor Xa. Lane 2: DsbC-K2S fusion protein. Lane 3: protein marker.

Thrombolytic activity assay of the recombinant K2S
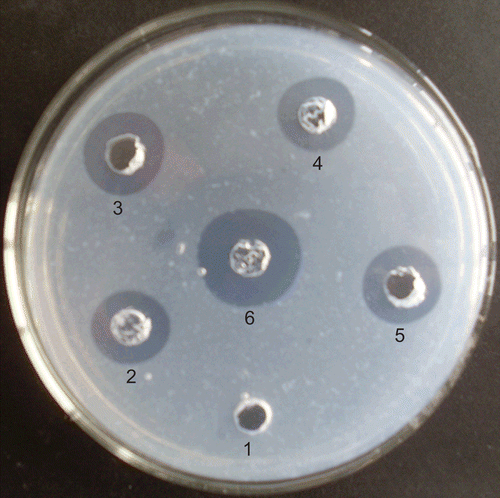
Ni2+-chelating affinity chromatography was performed again to purify the mature K2S from the cleaved protein solution. Then the thrombolytic activity of the recombinant K2S was determined using the fibrin plate assay. As shown in , the positive control tPA (spot 4), urokinase (spot 6), and the purified mature K2S (spots 3 and 5) showed a high thrombolytic activity, whereas no inhibition zone was seen around the spot of the expression product of pET40b (+) (spot 1). Furthermore, it is worth noting that the DsbC-K2S fusion protein (spot 2) also had significant thrombolytic activity. Comparing with tPA, the semiquantitative thrombolytic activity of K2S was 365.67 U/ml, and the activity of DsbC-K2S fusion protein was 323.50 U/ml. These results suggested that the DsbC chaperone could help the formation of the correct biological activity of K2S in the E. coli expression system.
Figure 6. Thrombolytic activity assay of the recombinant K2S. 1: protein product of pET40b (+) vector. 2: the DsbC-K2S fusion protein (125 μg). 3 and 5: K2S protein after Factor Xa cutting (50 μg). 4: tPA (10 U). 6: Urokinase (10 U).
Discussion
For the best application, it is desirable to express proteins in their soluble, active form. Solubility of a particular target protein is determined by a variety of factors. An alternative strategy to obtain active, soluble proteins is to construct a fusion protein with some peptides that enable export into the periplasm of E. coli, which is a more favorable environment for folding and disulfide-bond formation (CitationNakamoto & Bardwell, 2004; CitationInaba, 2009). The rate-limiting step in the oxidative folding of eukaryotic proteins in the periplasmic space of E. coli appears to be the formation and isomerization of disulfides bonds. Much evidence has revealed that the formation of disulfide bonds in E. coli is catalyzed by a complex machinery involving at least two soluble, periplasmic cysteine oxidoreductases (DsbA and DsbC), two membrane-bound enzymes (DsbB and DsbD), and cytoplasmic proteins (CitationMissiakas et al., 1995; CitationKe et al., 2006; CitationMessens & Collet, 2006; CitationInaba, 2008). In vitro, DsbA is a potent catalyst of protein cysteine oxidation, whereas DsbC exhibits disulfide isomerase activity. The membrane proteins DsbB and DsbD appear to be responsible for maintaining DsbA and DsbC, respectively, in the proper oxidative state for optimal function (CitationGoldstone et al., 2001; CitationIto & Inaba, 2008).
In the present study, K2S was modified to contain 357 of the 527 amino acids of the human tPA and remained nine disulfide bonds, which challenged the active production in E. coli. To solve this problem, we fused K2S with DsbC and its signal sequence. When using such recombinant vector for expression in E. coli, the fusion protein could be translocated into the periplasmic space and effectively catalyze the formation of disulfide bonds (CitationLiu & Wang, 2001). The results showed that the fusion protein DsbC-K2S was expressed as soluble form of ~40%. Furthermore, it was rather remarkable that in addition to the K2S protein cleaved from DsbC tag, the DsbC-K2S fusion protein also could exhibit significant fibrinolysis activity in vitro.
So far, to our best knowledge, few reports have described the production of soluble active K2S from E. coli. CitationQiu and his associates (1998) have successfully product active tPA in E. coli with co-overexpression of DsbC in individual vectors. CitationManosroi and his colleagues (2001) expressed the soluble periplasmic forms of K2S in E. coli with a phage-displayed vector. Compared to their studies, our results provided a more simple and effective strategy to obtain soluble active K2S in E. coli expression system. This research might contribute to the development of large-scale, bacterium-based K2S production.
Declaration of interest
This study was financially supported by the 863 (Hi-tech research and development program of China) program under contract NO. 2008AA10Z336.
References
- Adams HP Jr. (2003). Stroke: A vascular pathology with inadequate management. J Hypertens Suppl, 21, S3–S7.
- Baker WF Jr. (2005). Thrombolytic therapy: Current clinical practice. Hematol Oncol Clin North Am, 19, 147–81, vii.
- Gang J, Jiang CY, Du LX, Lu FP, Guo HJ, Li D, Liu HY. (2006). Cloning of reteplase gene and expression of the gene in Pichia methanolica. J South China Univ of Technol (Natural Science Edition) 12: 25–50.
- Goldstone D, Haebel PW, Katzen F, Bader MW, Bardwell JC, Beckwith J, Metcalf P. (2001). DsbC activation by the N-terminal domain of DsbD. Proc Natl Acad Sci USA, 98, 9551–9556.
- Inaba K. (2008). Protein disulfide bond generation in Escherichia coli DsbB-DsbA. J Synchrotron Radiat, 15, 199–201.
- Inaba K. (2009). Disulfide bond formation system in Escherichia coli. J Biochem, 146, 591–597.
- Ito K, Inaba K. (2008). The disulfide bond formation (Dsb) system. Curr Opin Struct Biol, 18, 450–458.
- Kalyan NK, Lee SG, Wilhelm J, Fu KP, Hum WT, Rappaport R, Hartzell RW, Urbano C, Hung PP. (1988). Structure-function analysis with tissue-type plasminogen activator. Effect of deletion of NH2-terminal domains on its biochemical and biological properties. J Biol Chem, 263, 3971–3978.
- Ke H, Zhang S, Li J, Howlett GJ, Wang CC. (2006). Folding of Escherichia coli DsbC: Characterization of a monomeric folding intermediate. Biochemistry, 45, 15100–15110.
- Larsen GR, Metzger M, Henson K, Blue Y, Horgan P. (1989). Pharmacokinetic and distribution analysis of variant forms of tissue-type plasminogen activator with prolonged clearance in rat. Blood, 73, 1842–1850.
- Liu X, Wang CC. (2001). Disulfide-dependent folding and export of Escherichia coli DsbC. J Biol Chem, 276, 1146–1151.
- Manosroi J, Tayapiwatana C, Götz F, Werner RG, Manosroi A. (2001). Secretion of active recombinant human tissue plasminogen activator derivatives in Escherichia coli. Appl Environ Microbiol, 67, 2657–2664.
- Messens J, Collet JF. (2006). Pathways of disulfide bond formation in Escherichia coli. Int J Biochem Cell Biol, 38, 1050–1062.
- Missiakas D, Schwager F, Raina S. (1995). Identification and characterization of a new disulfide isomerase-like protein (DsbD) in Escherichia coli. Embo J, 14, 3415–3424.
- Nakamoto H, Bardwell JC. (2004). Catalysis of disulfide bond formation and isomerization in the Escherichia coli periplasm. Biochim Biophys Acta, 1694, 111–119.
- Pennica D, Holmes WE, Kohr WJ, Harkins RN, Vehar GA, Ward CA, Bennett WF, Yelverton E, Seeburg PH, Heyneker HL, Goeddel DV, Collen D. (1983). Cloning and expression of human tissue-type plasminogen activator cDNA in E. coli. Nature, 301, 214–221.
- Qiu J, Swartz JR, Georgiou G. (1998). Expression of active human tissue-type plasminogen activator in Escherichia coli. Appl Environ Microbiol, 64, 4891–4896.
- Qureshi AI, Pande RU, Kim SH, Hanel RA, Kirmani JF, Yahia AM. (2002). Third generation thrombolytics for the treatment of ischemic stroke. Curr Opin Investig Drugs, 3, 1729–1732.
- Renatus M, Bode W, Huber R, Stürzebecher J, Prasa D, Fischer S, Kohnert U, Stubbs MT. (1997). Structural mapping of the active site specificity determinants of human tissue-type plasminogen activator. Implications for the design of low molecular weight substrates and inhibitors. J Biol Chem, 272, 21713–21719.
- Simpson D, Siddiqui MA, Scott LJ, Hilleman DE. (2006). Reteplase: a review of its use in the management of thrombotic occlusive disorders. Am J Cardiovasc Drugs, 6, 265–285.
- van Zonneveld AJ, Veerman H, Pannekoek H. (1986). Autonomous functions of structural domains on human tissue-type plasminogen activator. Proc Natl Acad Sci USA, 83, 4670–4674.
- Wooster MB, Luzier AB. (1999). Reteplase: A new thrombolytic for the treatment of acute myocardial infarction. Ann Pharmacother, 33, 318–324.