Abstract
Context: DNA topoisomerase I (topo I) is an essential enzyme which regulates the conformational changes in DNA topology by cleaving and rejoining DNA strands during normal cell growth. The inhibitors of topo I represent a major class of anticancer drugs. In our projects to isolate new anticancer agents from marine-derived fungi, secalonic acid D (SAD) with inhibitory activity on topo I was isolated from the fermentation broth of marine lichen-derived fungus Gliocladium sp. T31, which was collected from marine sediments in South Pole.
Objective: The inhibitory activity of SAD on topo I was investigated for the first time.
Materials and methods: The inhibitory effect of SAD on topo I was determined via in vitro supercoil relaxation assays and electrophoretic mobility shift assay (EMSA) using plasmid substrate, pBR322.
Results: SAD displays a considerable inhibition on topo I in a dose-dependent manner with the minimum inhibitory concentration (MIC) of 0.4 µM. Unlike the prototypic DNA topo I poison camptothecin (CPT), SAD inhibits the binding of topo I to DNA but does not induce the formation of topo I-DNA covalent complexes.
Discussion and conclusion: SAD is an excellent topo I inhibitor and thus a significantly potential anticancer candidate.
Introduction
DNA topoisomerases are essential enzymes which regulate the conformational changes in DNA topology by cleaving and realigning either one (topo I) or two (topo II) strands of DNA, thereby allowing DNA strands to pass through one another (CitationBugreev & Nevinsky, 2009; CitationGarcía-Estrada et al., 2010; CitationPommier et al., 2010). Considerable pharmacological investigations have been focused on DNA topoisomerase inhibitors, which have become a major class of anticancer drugs such as topotecan (CitationKollmannsberger et al., 1999), irinotecan (CitationBarth et al., 2010), fluoroquinolone (CitationBax et al., 2010).
In our projects to isolate novel anticancer agents from marine fungi, the ethyl acetate extract of fungus Gliocladium sp. T31 exhibited cytotoxicity against K562 cell line with the inhibition rate of 63% at a concentration of 100 µg/mL. In the exploration on the bioactive constituents of this fungal strain through a bioassay-guided isolation procedure, a main bioactive component, xanthone derivative secalonic acid D (SAD, ) was obtained. SAD, found as a potential inhibitor of protein kinases (CitationGanesh & Chada, 2000; CitationDhulipala et al., 2004), was commonly isolated from Aspergillus aculeatus and Blennoria sp. (CitationRaymond et al., 1977; CitationZhang et al., 2008). Our previous study has found that SAD blocks K562 cell proliferation at G0/G1 transition and has extent selectivity to different tumor cells, with strong cytotoxicity to K562, A549, and P388 cells and unnoticeable influence on BEL7402 cell (CitationRen et al., 2006). SAD contains symmetrical tetrahydroxanthone like diospyrin or other benzoxanthone derivatives, which belong to novel DNA topo I inhibitors (CitationTazi et al., 2005; CitationHee et al., 2010). Here, the inhibitory effects of SAD on DNA topo I was assessed experimentally for the first time in this research.
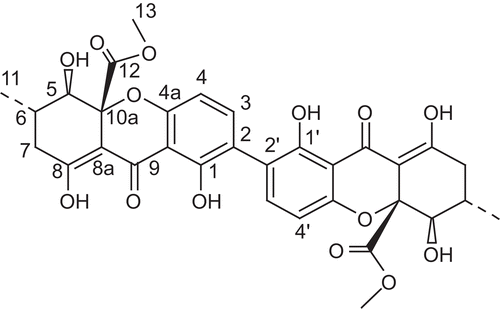
Figure 1. The structure of Secalonic acid D (SAD).
Materials and methods
General experimental procedures
Melting points were measured using a Yanaco MP-500D micro-melting point apparatus and were uncorrected. Optical rotations were obtained on a JASCO P-1020 digital polarimeter. Infrared (IR) spectra were obtained on a NICOLET NEXUS 470 spectrophotometer in KBr discs. 1H-, 13C-NMR and DEPT spectra and 2D-NMR data were recorded on a JEOL Eclips-600 spectrometer using TMS as internal standard and chemical shifts were recorded as δ values. ESI-MS was measured on a Q-TOF ULTIMA GLOBAL GAA076 LC mass spectrometer semiprepartive.
Fungus material
The fungal strain was separated from a marine lichen-derived fungus collected from South Pole (Zhong-shan Station, southern latitude 69°and east longitude 76°) in December 1999 and kept frozen at -20°C until fermentation. A voucher of this species (Gliocladium sp. T31) was deposited at the Natural Products Chemistry Laboratory, Institute of Marine Drugs and Food, Ocean University of China, China. The fungal strain was incubated in a hundred of Erlenmeyer flasks (500 mL type) on a rotatory shaker at 120 rpm and 28°C for 9 days, and each flask contained 150 mL of liquid medium with pH adjusted about 6.5, composed of mannitol 2%, glucose 1%, maltose 2%, monosodium glutamate 1%, corn syrup 0.1%, KH2PO4 0.05%, MgSO4·7H2O 0.03%, yeast extract 0.3%, and seawater.
Isolation of bioactive compounds
The fermentation broth and the mycelium were exhaustively extracted with EtOAc to afford a bioactive crude extract (32.9 g) with the inhibition rate of 63% at a concentration of 100 µg/mL, which was subjected by vacuum liquid chromatography over silica gel H using stepwise gradient elution with the mixtures of light petroleum ether-CHCl3-MeOH (1:8:1) to give three bioactive fraction A, B, and C, subsequently. Fraction A was recrystallized repeatedly in CHCl3-MeOH to obtain SAD (1) (503 mg), the yield in EtOAc extract was 1.53%.
SAD, pale yellow crystal, mp. 281–283°C, optical rotation of +61° (c0.11, CHCl3), IRνmax (KBr) cm−1: 3505 (chelated OH), 1735 (aliphatic ester C=O), 1610 (chelated O-hydroxyl C=O), 1585 (aromatic ring, C=C), 1432, 1232, 1061. 1H-NMR (600 MHz, DMSO-d6) δ: 13.60 (2H, brs, OH-8, OH-8’), 11.62 (2H, s, OH-1, OH-1’), 7.45 (2H, d, J = 8.42 Hz, H-3, H-3’), 6.63 (2H, d, J = 8.42 Hz, H-4, H-4’), 3.81 (2H, d, J = 9.51 Hz, H-5, H-5’), 3.61 (6H, s, H-13, H-13’), 2.65 (2H, dd, J = 8.42, 19.80 Hz, Ha-7, Ha-7’), 2.49 (2H, dd, J = 6.22, 19.80 Hz, Hb-7, Hb-7’), 2.31 (2H, m, H-6, H-6’), 1.03 (6H, d, J = 6.22 Hz, H-11, H-11’); 13C-NMR (150 MHz, DMSO-d6) δ: 186.6 (C-9, C-9’), 178.2 (C-8, C-8’), 170.0 (C-12, C-12’), 158.9 (C-1, C-1’), 158.5 (C-4a, C-4a’), 140.2 (C-3, C-3’), 117.3 (C-2, C-2’), 107.5 (C-4, C-4’), 106.3 (C-9a, C-9a’), 101.7 (C-8a, C-8a’), 85.2 (C-10a, C-10a’), 75.2 (C-5, C-5’), 52.6 (C-13, C-13’), 35.8 (C-7, C-7’), 29.9 (C-6, C-6’), 17.8 (C-11, C-11’).
Cell cultures
The human myeloid leukemia K562 cells were routinely maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) under a humidified atmosphere of 5% CO2 and 95% air at 37°C.
Analysis of DNA fragmentation
The method of DNA fragmentation analysis was used as described (CitationEng et al., 1988). Briefly, the cells were incubated at 50°C with lysis buffer containing 10 mM ethylenediaminetetraacetic acid (EDTA), 0.5% N-laurylsarcosine, and 0.5 mg/mL proteinase K in 50 mM Tris-HCl (pH 8.0), then RNase was added till the concentration of 0.25 mg/mL. DNA was extracted twice with phenol-chloroform and phenol-chloroform-isoamylalcohol in order. The extract was diluted with Tris EDTA (TE) buffer and centrifuged at 13,000 g for 10 min. The precipitated DNA was dissolved in TE buffer. The sample buffer and 0.1% ethidium bromide (EB) were subjected to electrophoresis on 1.5% agarose gel with Tris-borate EDTA buffer, and DNA was visualized under UV light.
Plasmid supercoil relaxation assay
Plasmid supercoil relaxation assays were carried out in typical procedure (CitationTopcu & Castora, 1995). Each reaction mixture had a total volume of 20 µL containing 50 mM Tris-HCl (pH 8.0), 50 mM EDTA, 0.5 mM dithiothreitol (DTT), 10% glycerol, 30 µg/mL bovine serum albumin, 2 µL test compound diluted with DMSO, 1U DNA topo I, and 0.2 µg pBR322 DNA. The reaction mixture was incubated at 37°C for 30 min, and the reaction was terminated by the addition of 5 µL stop solution containing 0.1% bromophenol blue, 4.5% sodium dodecyl sulfate (SDS), and 45% glycerol. The mixture was applied to 0.8% agarose gel and electrophoretically analyzed at 3.5 V/min, and the gel was stained with EB and photographed under UV light. One unit of the enzyme was defined as the minimal amount of activity required to relax 0.2 µg supercoiled pBR322 DNA under the conditions.
Analysis of topo I-DNA binding by EMSA
In the procedure of EMSA (CitationBoege et al., 1996), supercoiled pBR322 DNA was incubated in 20 mL relaxation topo I buffer with or without excess of topo I (400U) in the presence of test compounds at 37°C for 6 min. The reaction was started by the addition of DNA. The samples containing test compounds were assembled in order of compound, topo I, camptothecin (CPT). Samples were immediately loaded onto the 0.8% agarose gel in Tris-acetate-EDTA buffer with 1.0 µg/mL EB, then separated by electrophoresis at 2V/cm for 6 h, and photographed under UV light.
Results and discussion
The compound was obtained as a pale yellow crystal in CHCl3. Its molecular formula, C32H30O14, was determined by ESI-MS, 1H- and 13C-NMR spectra. The nuclear magnetic resonance (NMR) spectrum only displayed the presence of fifteen protons and sixteen carbons, implying that the compound was a symmetrical dimer. 1H-NMR, 13C-NMR, DEPT, and HMQC spectra revealed a half of the molecule, C16H15O7, possessing one conjugated ketone carbonyl δc 186.6 (C-9), one ester carbonyl 170.0 (C-12), seven quaternary carbons δc 178.2 (C-8), 158.9 (C-1), 158.5 (C-4a), 106.3 (C-9a), 101.7 (C-8a), 85.2 (C-10a), and 117.3 (C-2), two sp2 methines (δc 140.2, δH 7.45; δc 107.5, δH 6.63), one sp3 methines (δc 29.9, δH 2.31), one oxymethine (δc 75.2, δH 3.81), one methylene (δc 35.8, δH 2.65 and 2.49), one methoxyl (δc 52.6, δH 3.61), one methyl group (δc 17.8, δH 1.03), and two proton signals of chelated phenolic hydroxyl groups (δH 13.60 and 11.62). The connectivity of those groups could be elucidated by detailed analysis of its heteronuclear multiple quantum coherence (HMBC) spectrum and eventually to be identified as SAD (CitationSteyn, 1970). Furthermore, the compound shows a specific optical rotation of + 61° (c 0.11, CHCl3), compared with the previously reported data (CitationSteyn, 1970), similar to the reported value (+64°, c 0.14, CHCl3) for SAD, which further confirmed its structure (). It will suffice to indicate that the NMR, MS, and other physical data are in conformity with the previously published data (CitationSteyn, 1970).
The bioactive tetrahydroxanthone derivative SAD with potent cytotoxicity against K562, A549, P388, and BEL7402 cells was successfully isolated by bioassay-guided fractionation through repeated silica gel column chromatography and recrystallization from a crude EtOAc extract of the fungus G. sp. T31 (CitationRen et al., 2006). Whether SAD induced active apoptosis was further assessed for exploring potential antitumor agents from marine fungi. According to the diagnosis of the “DNA ladders” by agarose gel electrophoresis, the negative results in the DNA fragmentation analysis suggested that the cell death caused by various levels of SAD was unrelated to apoptosis.
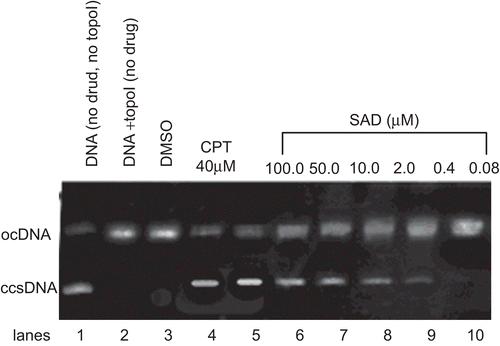
More than ten xanthone derivatives have been identified previously as DNA topoisomerase inhibitors (CitationWebb & Ebeler, 2004; CitationMa et al., 2005). SAD to interfere with mammalian DNA topo I was investigated via in vitro supercoil relaxation assays using plasmid substrate, pBR322, with the well-known topo I inhibitor CPT as positive control. The interference in the supercoil relaxing activity of topo I in the presence of SAD with various concentrations is shown in , where the ccsDNA (, lane 1) was fully relaxed by the enzyme (, lane 2). The organic solvent of DMSO exerted no detectable inhibition on the DNA topo I activity under the identical experimental conditions (, lane 3). Relaxation of ccs plasmid substrate, pBR322, was also inhibited upon the incubation with CPT (, lane 4) and SAD (, lane 5 ~ 10). Both CPT and SAD strongly inhibited the DNA relaxation activity of mammalian topo I, where the inhibition was diminished at the the minimum inhibitory concentration (MIC) of 0.4 µM, reflected by the accumulation of faster migrating ccs pBR322 (, lane 9).
Figure 2. Agarose gel photograph of the plasmid supercoil relaxation assay in the presence of DMSO, camptothecin (CPT), and various concentrations of SAD. Lane 1, pBR322 plasmid DNA without enzyme; lane 2, supercoil relaxation with 1 unit of DNA topoisomerase I; lane 3, same as lane 2 in the presence of DMSO; lane 4, same as lane 2 in the presence of CPT at the concentration of 40 µM; lanes 5–10, same as lane 2 in the presence of decreasing concentrations of SAD (100 to 0.08 µM). This experiment was repeated three times with similar results.
In order to assess whether SAD interferes in the binding of topo I to DNA, we performed EMSA between supercoiled pBR322 DNA and topo I using CPT as positive control, which inhibits the ligase activity and does not interfere with the binding of the enzyme to DNA. The Results (shown in ) clearly indicated that SAD, unlike CPT, hampered the binding of topo I to DNA at the concentration of 40 µM.
Figure 3. Representative image of the agarose gel electrophoresis for EMSA between pBR322 DNA and topo I. Supercoiled pBR322 was incubated without excess of topo I (100 U) in the presence of the test compound at 37°C for 6 min. The reaction was started by addition of DNA. Samples were assembled in the order compound, topo I, topo I inhibitor. This experiment was repeated three times with similar results.

Conclusion
A potent antitumor tetrahydroxanthone dimer SAD was obtained from marine lichen-derived fungus G. sp. T31 by a bioassay-guided separation procedure. In this study, we assessed the ability of SAD to inhibit mammalian DNA topo I via in vitro plasmid supercoil relaxation assay and EMSA. The results clearly indicate that SAD is a novel inhibitor of DNA topo I. SAD exerts a different mechanism of action from CPT, CPT does not interfere with the formation of the complex DNA-topo I, whereas SAD significantly hamper the binding of the enzyme to DNA. The data add to the understanding that SAD is a DNA topo I inhibitor rather than a poison. This is the first study reporting an inhibitory effect of SAD on mammalian DNA topo I. In summary, the ability of SAD to inhibit the proliferation of several tumor cells as well as to interfere with DNA topo I suggested that its potential for inhibiting human tumor cells needs to be studied.
Acknowlegements
We are grateful to Prof. Tian Li (The first Institute of Oceanography, State Oceanic Administration) for the identification of marine lichen and the isolated fungus Gliocladium sp. T31. This work was financially supported by the Fund of Key laboratory of Marine Drug Chinese Ministry of Education [KLMD(OUC)200409] and Chinese National Natural Science Fund (No.30472136&30470196). The anti-tumor assay was performed at the Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
Declaration of interest
The authors report no conflicts of interest.
Related Research Data
References
- Barth SW, Briviba K, Watzl B, Jäger N, Marko D, Esselen M. (2010). In vivo bioassay to detect irinotecan-stabilized DNA/topoisomerase I complexes in rats. Biotechnol J, 5, 321–327.
- Bax BD, Chan PF, Eggleston DS, Fosberry A, Gentry DR, Gorrec F, Giordano I, Hann MM, Hennessy A, Hibbs M, Huang J, Jones E, Jones J, Brown KK, Lewis CJ, May EW, Saunders MR, Singh O, Spitzfaden CE, Shen C, Shillings A, Theobald AJ, Wohlkonig A, Pearson ND, Gwynn MN. (2010). Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature, 466, 935–940.
- Boege F, Straub T, Kehr A, Boesenberg C, Christiansen K, Andersen A. (1996). Selected novel flavones inhibit the DNA binding or the DNA relegation step of eukaryotic topoisomerase I. J Biol Chem, 271 (4): 2262–2270.
- Bugreev DV, Nevinsky GA. (2009). Structure and mechanism of action of type IA DNA topoisomerases. Biochemistry (Mosc), 74, 1467–1481.
- Dhulipala VC, Hanumegowda UM, Balasubramanian G, Reddy CS. (2004). Relevance of the palatal protein kinase A pathway to the pathogenesis of cleft palate by secalonic acid D in mice. Toxicol Appl Pharmacol, 194, 270–279.
- Eng WK, Faucette L, Johnson RK, Sternglanz R. (1988). Evidence that DNA topoisomerase I is necessary for the cytotoxic effects of camptothecin. Mol Pharmacol, 34, 755–760.
- Ganesh B, Chada SR. (2000). Novel mechanism of protein kinase C inhibition involving the pseudosubstrate region by secalonic acid D in vitro. Toxicol Appl Pharmacol, 163, 86–93.
- García-Estrada C, Prada CF, Fernández-Rubio C, Rojo-Vázquez F, Balaña-Fouce R. (2010). DNA topoisomerases in apicomplexan parasites: Promising targets for drug discovery. Proc Biol Sci, 277, 1777–1787.
- Hee JC, Mi JJ, Sangwook W, Jungsook K, Eung SL, Youngjoo K, Younghwa N. (2010). New benzoxanthone derivatives as topoisomerase inhibitors and DNA cross-linkers. Bioorg Med Chem, 18, 1010–1017.
- Kollmannsberger C, Mross K, Jakob A, Kanz L, Bokemeyer C. (1999). Topotecan - A novel topoisomerase I inhibitor: Pharmacology and clinical experience. Oncology, 56, 1–12.
- Ma J, Jones SH, Marshall R, Wu X, Hecht SM. (2005). DNA topoisomerase I inhibitors from Rinorea anguifera. Bioorg Med Chem Lett, 15, 813–816.
- Pommier Y, Leo E, Zhang H, Marchand C. (2010). DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 17 (5): 421–33.
- Raymond A, George B, Brunhilde K, Arnold LD. (1977). Secalonic acid D and F are toxic metabolites of Aspergillus aculeatus. J Org Chem, 42, 352–353.
- Ren H, Tian L, Gu Q, Zhu W. (2006). Secalonic acid D; A cytotoxic constituent from marine lichen-derived fungus Gliocladium sp. T31. Arch Pharm Res, 29, 59–63.
- Steyn PS. (1970). The isolation, structure and absolute configuration of secalonic acid D, the toxic metabolite of Penicillium oxalicum. Tetrahedron, 26, 51–57.
- Tazi J, Bakkour N, Soret J, Zekri L, Hazra B, Laine W, Baldeyrou B, Lansiaux A, Bailly C. (2005). Selective inhibition of topoisomerase I and various steps of spliceosome assembly by diospyrin derivatives. Mol Pharmacol, 67, 1186–1194.
- Topcu Z, Castora FJ. (1995). Mammalian mitochondrial DNA topoisomerase I preferentially relaxes supercoils in plasmids containing specific mitochondrial DNA sequences. Biochim Biophys Acta, 1264, 377–387.
- Webb MR, Ebeler SE. (2004). Comparative analysis of topoisomerase IB inhibition and DNA intercalation by flavonoids and similar compounds: Structural determinates of activity. Biochem J, 384, 527–541.
- Zhang W, Krohn K, Flörke U, Pescitelli G, Di Bari L, Antus S, Kurtán T, Rheinheimer J, Draeger S, Schulz B; Zia-Ullah. (2008). New mono- and dimeric members of the secalonic acid family: Blennolides A-G isolated from the fungus Blennoria sp.. Chemistry, 14, 4913–4923.