Abstract
Context: As an inhibitor of CYP2C9, CYP2D6 and P-gp, myricetin might affect the bioavailability of carvedilol when myricetin and carvedilol are used concomitantly for the prevention or therapy of cardiovascular diseases as a combination therapy. However, the effect of myricetin on the pharmacokinetics of carvedilol has not been reported in vivo.
Objective: This study investigated the effects of myricetin on the pharmacokinetics of carvedilol after oral or intravenous administration of carvedilol in rats.
Materials and methods: Carvedilol was administered orally or intravenously with or without oral administration of myricetin to rats.
Results: The effects of myricetin on P-gp, CYP2C9 and 2D6 activity were evaluated. Myricetin inhibited CYP2C9 and CYP2D6 enzyme activity with IC50 of 13 and 57 μM, respectively. In addition, myricetin significantly enhanced the cellular accumulation of rhodamine-123 in MCF-7/ADR cells overexpressing P-gp. Compared with the control group, the AUC was significantly increased by 52.0–85.1%, and the Cmax was significantly increased by 93.1–133.4% in the presence of myricetin after oral administration of carvedilol. Consequently, the relative bioavailability of carvedilol was increased by 1.17- to 1.85-fold and the absolute bioavailability of carvedilol in the presence of myricetin was increased by 18.1–86.4%. Tmax was significantly decreased.
Discussion and conclusion: The enhanced oral bioavailability of carvedilol may result from both inhibition of CYP2C9 or CYP2D6-mediated metabolism and P-gp-mediated efflux of carvedilol in small intestine and/or in liver by myricetin rather than reducing renal elimination. Concomitant use of myricetin or myricetin-containing dietary supplements with carvedilol will require close monitoring for potential drug interactions.
Introduction
Carvedilol {(±)-1-carbazol-4-yloxy)-3-[[2-(omethoxyphenoxy)ethyl]-amino]-2-propanol} is an antihypertensive and antianginal compound which combines nonselective β-adrenoceptor blocking and vasodilator properties with intrinsic sympathomimetic activity (Citationvon Mollendorff et al., 1986; CitationFrishman, 1998). It is used to treat systemic arterial hypertension (CitationCournot et al., 1992; CitationLund-Johansen et al., 1992) and congestive heart failure (CitationDasGupta et al., 1991). It is also used to reduce mortality in patients with left ventricular dysfunction following myocardial infarction (CitationAbshagen, 1987). Carvedilol is well absorbed from the gastrointestinal tract, but is subject to considerable first-pass metabolism in the intestinal and/or liver (CitationMcTavish et al., 1993; CitationMorgan, 1994). Carvedilol is more than 98% bound to plasma proteins. Carvedilol is metabolized by both oxidation and conjugation pathways in the liver into some metabolites (CitationNeugebauer et al., 1987; CitationNeugebauer & Neubert, 1991). The oxidation pathways are mainly catalyzed by CYP2C9 and CYP2D6 enzymes in human being (CitationMcTavish et al., 1993; CitationMorgan, 1994; CitationOldham & Clarke, 1997), and then CYP2D6 is responsible for the formation of 4-hydroxy carvedilol and 5-hydroxy carvedilol, and both metabolites are excreted into urine (CitationNeugebauer & Neubert, 1991). Carvedilol is also a substrate of P-gp (CitationBart et al., 2005). Since carvedilol is a substrate of both CYP enzymes and P-gp, the modulation of CYP and P-gp activities may cause significant changes in the pharmacokinetic profile of carvedilol.
Flavonoids represent a group of phytochemicals that are produced by various plants in high quantities (CitationDixon & Steele, 1999). They exhibit a wide range of beneficial biological activities including antioxidative, radical scavenging, antiatherosclerotic, antitumor and antiviral effects (CitationNijveldt et al., 2001). Myricetin is a naturally occurring flavonol, a flavonoid found in several foods including onions, berries and grapes as well as red wine (CitationHakkinen et al., 1999). Myricetin plays an important role in anti-hemorrhagic potential (CitationNishijima et al., 2009), anti-myocardial infarction (CitationTiwari et al., 2009), and anti-hypertension (CitationXue et al., 2008).
Citationvon Moltke et al. (2004) reported that myricetin inhibits human CYP3A4 and 2C9 while CitationVáclavíková et al. (2003) found that myricetin inhibits human CYP3A4 and 2C8. Thus, the inhibitory effects of myricetin against human CYP enzymes remain somewhat controversial. Myricetin is an inhibitor of P-gp in the KB/MDR cell line (CitationKitagawa et al., 2005), but the inhibitory effect of myricetin against P-gp is otherwise ambiguous. Therefore, we re-evaluated the inhibition of CYP enzyme activity and P-gp activity by myricetin using CYP inhibition assays and rhodamine-123 retention assays in P-gp-overexpressing MCF-7/ADR cells.
As an inhibitor of CYP2C9, CYP2D6 and P-gp, myricetin might affect the bioavailability and pharmacokinetics of carvedilol when myricetin and carvedilol are used concomitantly for the prevention or therapy of cardiovascular diseases as a combination therapy. However, the effect of myricetin on the pharmacokinetics of carvedilol has not been reported in vivo. This study focused on the effect of myricetin on the bioavailability and pharmacokinetics of carvedilol after oral and intravenous administrarion of carvedilol in rats.
Materials and methods
Chemicals and apparatus
Carvedilol, myricetin and nimodipine (an internal standard for high-performance liquid chromatograph (HPLC) analysis for carvedilol) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). HPLC grade acetonitrile was acquired from Merck Co. (Darmstadt, Germany). Other chemicals for this study were of reagent grade.
Apparatuses used in this study were a HPLC equipped with a Waters 1515 isocratic HPLC Pump, a Waters 717 plus autosampler and a Waters™ 474 scanning fluorescence detector (Waters Co., Milford, MA, USA), a HPLC column temperature controller (Phenomenex Inc., CA, USA), a Bransonic® Ultrasonic Cleaner (Branson Ultrasonic Co., Danbury, CT, USA), a vortex-mixer (Scientific Industries Co., NY, USA) and a high-speed micro centrifuge (Hitachi Co., Tokyo, Japan).
Animal experiments
Male Sprague–Dawley rats of 7–8 weeks of age (weighing 270–300 g) were purchased from Dae Han Laboratory Animal Research Co. (Choongbuk, Republic of Korea) and given free access to a commercial rat chow diet (No. 322-7-1; Superfeed Co., Gangwon, Republic of Korea) and tap water ad libitum. The animals were housed (two rats per cage) in a clean room maintained at a temperature of 22 ± 2°C and relative humidity of 50–60%, with 12 h light/dark cycles. The rats were acclimated under these conditions for at least 1 week. All animal studies were performed in accordance with the “Guiding Principles in the Use of Animals in Toxicology” adopted by the Society of Toxicology (USA) and the Animal Care Committee of Chosun University (Gwangju, Republic of Korea) approved the protocol of this animal study. The rats were fasted for at least 24 h prior to beginning the experiments and had free access to tap water. Each animal was anaesthetized lightly with ether. The left femoral artery and vein were cannulated using polyethylene tubing (SP45, I.D. 0.58 mm, O.D. 0.96 mm; Natsume Seisakusho Co. LTD., Tokyo, Japan) for blood sampling and i.v. injection, respectively.
Oral and intravenous administration of carvedilol
The rats were divided into eight groups (n = 6, each): an oral group (3 mg/kg of carvedilol dissolved in water; homogenized at 36°C for 30 min; 3.0 mL/kg) without (control) or with 0.3, 1.5 or 6 mg/kg of oral myricetin, and an i.v. group (1 mg/kg of carvedilol, dissolved in 0.9% NaCl solution; homogenized at 36°C for 30 min; 1.5 mL/kg) without (control) or with 0.3, 1.5 or 6 mg/kg of oral myricetin. Myricetin was orally administered 30 min prior to oral or intravenous administration of carvedilol. Oral carvedilol was administered through a feeding tube, and carvedilol for i.v. administration was injected through the femoral vein within 0.5 min. A 0.4 mL blood sample was collected into heparinized tubes from the femoral artery at 0, 0.1, 0.25, 0.5, 1, 2, 4, 8, 12 and 24 h after intravenous infusion and at 0, 0.25, 0.5, 1, 2, 4, 8, 12 and 24 h after oral administration. The blood samples were centrifuged (13,000 rpm, 5 min), and the plasma samples were stored at−40°C until HPLC analysis of carvedilol. Rats were infused with approximately 1 mL of whole blood collected from untreated rats via the femoral artery at 1/2, 2 and 8 h to replace the blood loss due to blood sampling.
HPLC assay
The plasma concentrations of carvedilol were determined by the HPLC assay method reported by CitationZarghi et al. (2007). Briefly, 50 μL of nimodipine (20 μg/mL dissolved in methanol; an internal standard) and 0.5 mL of acetonitrile were added to a 0.2 mL aliquot of the plasma in a 2.0 mL polypropylene microtube. The mixture was then stirred for 10 min and centrifuged (13,000 rpm, 10 min). A 0.5 mL aliquot of the organic layer was transferred to a clean test tube and evaporated under a gentle stream of nitrogen gas at 35°C. The residue was reconstituted in a 150 μL of the mobile phase and centrifuged (13,000 rpm, 5 min). The resulting mixture was then vigorously vortex-mixed for 5 min and centrifuged at 13,000 rpm for 5 min. A 50 μL aliquot of the supernatant was injected into the HPLC system. Chromatographic separations were achieved using a Chromolith Performance (RP-18e, 100 mm × 4.6 mm) column from Merck (Darmstadt, Germany). The mobile phase consisted of 0.01 M disodium hydrogen phosphate (pH 3.5, adjusted with phosphoric acid)-acetonitrile (75.7:24.3, v/v). The flow rate of the mobile phase was maintained at 2.0 mL/min. Chromatography was performed at of 25°C, which was regulated by an HPLC column temperature controller. The fluorescence detector was operated at an excitation wavelength of 240 nm with an emission wavelength of 340 nm. The retention times at a flow rate of 2 mL/min were as follows: carvedilol at 8.076 min internal standard at 9.305 min. The lower limit of quantification for carvedilol in rat plasma was 10 ng/mL. The coefficient of the variation of carvedilol was less than 14.3%.
CYP 2C9 and 2D6 inhibition assay
The assays of inhibition on CYP2C9 and 2D6 enzyme activities were performed in a multiwell plate using a CYP inhibition assay kit (GENTEST, Woburn, MA) as described by CitationCrespi et al. (1997). Briefly, human CYP enzymes were obtained from baculovirus-infected insect cells. CYP substrates (7-MFC for CYP2D6) were incubated with or without test compounds in the enzyme/substrate contained buffer consisting of 1 pmol of P450 enzyme and NADPH generating system (1.3 mM NADP, 3.54 mM glucose 6-phosphate, 0.4 U/mL glucose 6-phosphate dehydrogenase and 3.3 mM MgCl2) in a potassium phosphate buffer (pH 7.4). Reactions were terminated by adding stop solution after 45 min incubation. Metabolite concentrations were measured by spectrofluorometer (Molecular Device, Sunnyvale, CA) set at an excitation wavelength of 409 nm and an emission wavelength of 530 nm. Positive control (2 μM sulfaphenazole for CYP2C9) was run on the same plate and produced 99% inhibition. All experiments were performed in duplicate, and results are expressed as percentage of inhibition.
Rhodamine-123 retention assay
The procedures used for Rho-123 retension assay were similar to a reported method (CitationHan et al., 2008). MCF-7/ADR cells were seeded in 24-well plates. At 80% confluence, the cells were incubated in FBS-free DMEM for 18 h. The culture medium was changed to Hanks’ balanced salt solution and the cells were incubated at 37°C for 30 min. After incubation of the cells with 20 μM rhodamine-123 for 90 min, the medium was completely removed. The cells were then washed three times with ice-cold phosphate buffer (pH 7.0) and lysed in lysis buffer. The rhodamine-123 fluorescence in the cell lysates was measured using excitation and emission wavelengths of 480 and 540 nm, respectively. Fluorescence values were normalized to the total protein content of each sample and presented as the ratio to controls.
Pharmacokinetic analysis
The plasma concentration data were analyzed by the noncompartmental method using WinNonlin software version 4.1 (Pharsight Co., Mountain View, CA, USA). The elimination rate constant (Kel) was calculated by log-linear regression of carvedilol concentration data during the elimination phase, and the terminal half-life (t1/2) was calculated by 0.693/Kel. The peak concentration (Cmax) and the time to reach peak concentration (Tmax) of carvedilol in plasma were obtained by visual inspection of the data from the concentration-time curve. The area under the plasma concentration-time curve (AUC0–t) from time zero to the time of last measured concentration (Clast) was calculated by the linear trapezoidal rule. The AUC zero to infinity (AUC0–∞) was obtained by the addition of AUC0–t and the extrapolated area determined by Clast/Kel. Total body clearance (CL/F) was calculated by Dose/AUC. The absolute bioavailability (AB) of carvedilol was calculated by AUCoral/AUCiv × Dosei.v./Doseoral × 100, and the relative bioavailability (RB) of carvedilol was estimated by AUCwith myricetin/AUCcontrol × 100.
Statistical analysis
All mean values are presented with their standard deviation (mean ± SD). Statistical analysis was conducted using one-way ANOVA followed by a posteriori testing with Dunnett’s correction. Differences were considered significant at a level of p < 0.05.
Results
Inhibition of CYP2C9 and 2D6
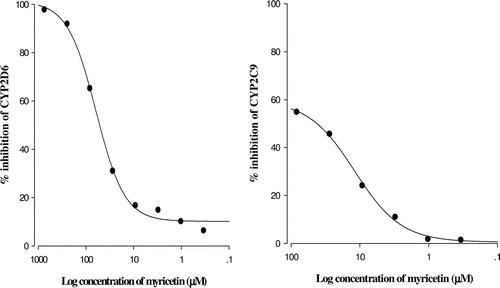
The inhibitory effect of myricetin on CYP2C9 and CYP2D6 activity is shown in . Myricetin inhibited CYP2C9 and CYP2D6 activity in a concentration-dependent manner, and the 50% inhibition concentration (IC50) values of myricetin on CYP2C9 and CYP2D6 activity were 13 and 57 μM.
Figure 1. Inhibitory effect of myricetin on CYP2C9 and 2D6 activity. All experiments were performed in duplicate, and results are expressed as the percent of inhibition. The IC50 values of myricetin on CYP2C9 and 2D6 activity are 13 and 59 μM, respectively.
Rhodamine-123 retention assay
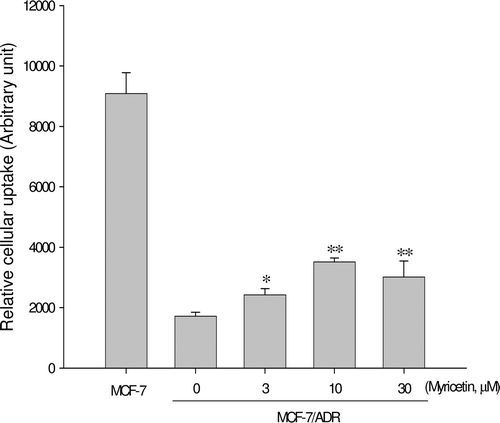
As shown in , accumulation of rhodamine-123, a P-gp substrate, was reduced in MCF-7/ADR cells overexpressing P-gp compared to that in MCF-7 cells lacking P-gp. The concurrent use of myricetin enhanced the cellular uptake of rhodamine 123 in a concentration-dependent manner and showed statistically significant (p < 0.01) increase at concentrations ranging from 3–10 μM. This result suggests that myricetin significantly inhibits P-gp activity.
Figure 2. Rhodamine-123 retention. MCF-7/ADR cells were preincubated with myricetin for 24 h, and incubation of MCF-7/ADR cells with 20 μM R-123 for 90 min. Data represents mean ± SD of 6 separate samples (significant versus the control MCF-7 cells; *p < 0.05, **p < 0.01).
Effects of myricetin on plasma concentrations after oral administration
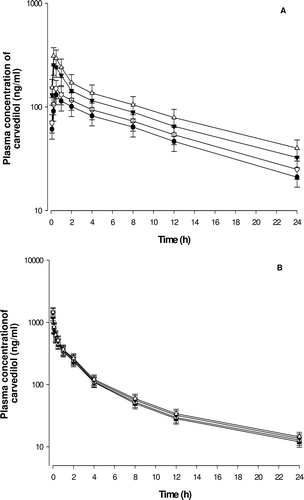
The mean plasma concentration-time profiles of oral carvedilol in the presence or absence of myricetin are illustrated in . The mean pharmacokinetic parameters of carvedilol are also summarized in . shows the plasma concentration-time profiles of carvedilol after oral administration of 3 mg/kg of carvedilol in rats with or without myricetin (0.3, 1.5 or 6 mg/kg), and the pharmacokinetic parameters of oral carvedilol are summarized in . The area under the plasma concentration-time curve (AUC) was significantly (1.5 mg/kg, p < 0.05; 6 mg/kg, p < 0.01) increased by 52.0–85.1%, and the peak concentration (Cmax) was significantly (1.5 mg/kg, p < 0.05; 6 mg/kg, p < 0.01) increased by 93.1–133.4% in the presence of myricetin after oral administration of carvedilol. Consequently, the relative bioavailability (RB) of carvedilol was increased by 1.17- to 1.85-fold, and the absolute bioavailability (AB) of carvedilol in the presence of myricetin was increased by 18.1–86.4%. Tmax was significantly (1.5 mg/kg and 6 mg/kg, p < 0.05) decreased. However, there were no significant changes in the half-life (t1/2) of carvedilol in the presence of myricetin.
Table 1. Pharmacokinetic parameters of carvedilol after oral and intravenous administration of carvedilol in the presence or absence of myricetin to rats.
Figure 3. Mean plasma concentration–time profiles of carvedilol after oral (3 mg/kg, A) and intravenous (1 mg/kg, B) administration of carvedilol without (•) or with 0.3 mg/kg (○), 1.5 mg/kg (▾) and 6 mg/kg (▵) of myricetin to rats. Bars represent the standard deviation (n = 6).
Effects of myricetin on plasma concentrations after i.v. administration
The mean plasma concentration-time profiles of i.v. carvedilol in the presence or absence of myricetin are illustrated in . The mean pharmacokinetic parameters of carvedilol are also summarized in . shows the plasma concentration-time profiles of carvedilol after i.v. (1 mg/kg) administration without or with of myricetin (0.3, 1.5 or 6 mg/kg) to rats. As shown in , myricetin did not significantly change the pharmacokinetic parameters of i.v. administration of carvedilol.
Discussion
CYPs enzymes significantly contribute to the first-pass metabolism and oral bioavailability of many drugs. The first-pass metabolism of compounds in the intestine limits absorption of toxic xenobiotics and may ameliorate side effects. Moreover, induction or inhibition of intestinal CYPs may be responsible for significant drug and drug interactions when one agent decreases or increases the bioavailability of a concurrently administered drug (CitationKaminsky & Fasco, 1991).
Based on the broad overlap in the substrate specificities as well as co-localization in the small intestine, the primary site of absorption for orally administered drugs, CYP3A4, CYP2C9 and P-gp have been recognized as a concerted barrier to the drug absorption (CitationCummins et al., 2002; CitationBenet et al., 2003). Therefore, inhibitors against both CYP2C9 and P-gp should have a great impact on the bioavailability of many drugs, where CYP2C9 metabolism as well as P-gp mediated efflux is the major barrier to the systemic availability.
The inhibitory effect of myricetin against CYP2C9-mediated metabolism was confirmed by the employment of recombinant CYP2C9 enzyme. As shown in , myricetin exhibited inhibitory effect against CYP2D6 and CYP2C9-mediated metabolism with IC50 of 57 and 13 μM, respectively. Furthermore, the cell-based assay using rhodamine-123 indicated that myricetin (30 μM) significantly (p < 0.01) inhibited P-gp-mediated drug efflux (). These results are consistent with the previous report (Citationvon Moltke et al., 2004; CitationKitagawa et al., 2005). Therefore, the pharmacokinetic characteristics of carvedilol were evaluated in the absence and presence of myricetin in rats. Human CYP2C9 and 3A4 and rat CYP2C11 and 3A1 have 77 and 73% protein homology, respectively (CitationLewis, 1996). Rats were selected as an animal model in this study to evaluate the potential pharmacokinetic interactions mediated by CYP2C9, although there may be some difference in enzyme activity between a rat and a human (CitationCao et al., 2006). Therefore, myricetin might possibly increase absorption of carvedilol in the intestine through the inhibition of P-gp and CYP2C9.
In addition to its extensive metabolism by CYP2C9, carvedilol appeared to be the substrate of P-gp, suggesting that P-gp and CYP2C9 should act synergistically to limit the oral bioavailability of carvedilol (CitationSaeki et al., 1993). Studies on drug interactions with grapefruit juice have provided much understanding of the role of intestinal CYP450 in the absorption of orally administered drugs. CYP2C9 is the predominant P450 present in the small intestine (CitationKolars et al., 1992). Orally administered carvedilol is a substrate for CYP2C9-mediated metabolism and P-gp-mediated efflux. The enhanced oral bioavailability of carvedilol might be due both to inhibition of P-gp efflux pump in the small intestine and CYP2C9 metabolism of carvedilol in the intestine and/or liver by myricetin.
Conclusions
The increased bioavailability of carvedilol might be due both to inhibition of P-gp efflux in the intestine and CYP2C9, 2D6-mediated metabolism in the intestine and/or in liver rather than renal elimination by myricetin. Therefore, concomitant use of myricetin or myricetin-containing dietary supplements with carvedilol will require close monitoring for potential drug interactions.
Declaration of interest
The authors declare no conflicts of interest.
References
- Abshagen U. (1987). A new molecule with vasodilating and beta-adrenoceptor blocking properties. J Cardiovasc Pharmacol, 10 Suppl 11, S23–S32.
- Bart J, Dijkers EC, Wegman TD, de Vries EG, van der Graaf WT, Groen HJ, Vaalburg W, Willemsen AT, Hendrikse NH. (2005). New positron emission tomography tracer [(11)C]carvedilol reveals P-glycoprotein modulation kinetics. Br J Pharmacol, 145, 1045–1051.
- Benet LZ, Cummins CL, Wu CY. (2003). Transporter-enzyme interactions: implications for predicting drug-drug interactions from in vitro data. Curr Drug Metab, 4, 393–398.
- Cao X, Gibbs ST, Fang L, Miller HA, Landowski CP, Shin HC, Lennernas H, Zhong Y, Amidon GL, Yu LX, Sun D. (2006). Why is it challenging to predict intestinal drug absorption and oral bioavailability in human using rat model. Pharm Res, 23, 1675–1686.
- Cournot A, Lim C, Duchier J, Safar M. (1992). Hemodynamic effects of carvedilol after acute oral administration in hypertensive and normal subjects. J Cardiovasc Pharmacol, 19 Suppl 1, S35–S39.
- Crespi CL, Miller VP, Penman BW. (1997). Microtiter plate assays for inhibition of human, drug-metabolizing cytochromes P450. Anal Biochem, 248, 188–190.
- Cummins CL, Jacobsen W, Benet LZ. (2002). Unmasking the dynamic interplay between intestinal P-glycoprotein and CYP3A4. J Pharmacol Exp Ther, 300, 1036–1045.
- DasGupta P, Broadhurst P, Lahiri A. (1991). The effects of intravenous carvedilol, a new multiple action vasodilatory beta-blocker, in congestive heart failure. J Cardiovasc Pharmacol, 18 Suppl 4, S12–S16.
- Dixon RA, Steele CL. (1999). Flavonoids and isoflavonoids - a gold mine for metabolic engineering. Trends Plant Sci, 4, 394–400.
- Frishman WH. (1998). Carvedilol. N Engl J Med, 339, 1759–1765.
- Häkkinen SH, Kärenlampi SO, Heinonen IM, Mykkänen HM, Törrönen AR. (1999). Content of the flavonols quercetin, myricetin, and kaempferol in 25 edible berries. J Agric Food Chem, 47, 2274–2279.
- Hampton JR. (1996). Beta-blockers in heart failure–the evidence from clinical trials. Eur Heart J, 17 Suppl B, 17–20.
- Han CY, Cho KB, Choi HS, Han HK, Kang KW. (2008). Role of FoxO1 activation in MDR1 expression in adriamycin-resistant breast cancer cells. Carcinogenesis, 29, 1837–1844.
- Kaminsky LS, Fasco MJ. (1991). Small intestinal cytochromes P450. Crit Rev Toxicol, 21, 407–422.
- Kitagawa S, Nabekura T, Takahashi T, Nakamura Y, Sakamoto H, Tano H, Hirai M, Tsukahara G. (2005). Structure-activity relationships of the inhibitory effects of flavonoids on P-glycoprotein-mediated transport in KB-C2 cells. Biol Pharm Bull, 28, 2274–2278.
- Kolars JC, Schmiedlin-Ren P, Schuetz JD, Fang C, Watkins PB. (1992). Identification of rifampin-inducible P450IIIA4 (CYP3A4) in human small bowel enterocytes. J Clin Invest, 90, 1871–1878.
- Lewis DFV. (1996). Cytochromes P450: Structure, Function and Mechanism. Bristol, England, Taylor & Francis Publisher and Distributor.
- Lund-Johansen P, Omvik P, Nordrehaug JE, White W. (1992). Carvedilol in hypertension: effects on hemodynamics and 24-hour blood pressure. J Cardiovasc Pharmacol, 19 Suppl 1, S27–S34.
- McTavish D, Campoli-Richards D, Sorkin EM. (1993). Carvedilol. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy. Drugs, 45, 232–258.
- Morgan T. (1994). Clinical pharmacokinetics and pharmacodynamics of carvedilol. Clin Pharmacokinet, 26, 335–346.
- Neugebauer G, Akpan W, von Möllendorff E, Neubert P, Reiff K. (1987). Pharmacokinetics and disposition of carvedilol in humans. J Cardiovasc Pharmacol, 10 Suppl 11, S85–S88.
- Neugebauer G, Neubert P. (1991). Metabolism of carvedilol in man. Eur J Drug Metab Pharmacokinet, 16, 257–260.
- Nijveldt RJ, van Nood E, van Hoorn DE, Boelens PG, van Norren K, van Leeuwen PA. (2001). Flavonoids: a review of probable mechanisms of action and potential applications. Am J Clin Nutr, 74, 418–425.
- Nishijima CM, Rodrigues CM, Silva MA, Lopes-Ferreira M, Vilegas W, Hiruma-Lima CA. (2009). Anti-hemorrhagic activity of four Brazilian vegetable species against Bothrops jararaca venom. Molecules, 14, 1072–1080.
- Oldham HG, Clarke SE. (1997). In vitro identification of the human cytochrome P450 enzymes involved in the metabolism of R(+)- and S(-)-carvedilol. Drug Metab Dispos, 25, 970–977.
- Saeki T, Ueda K, Tanigawara Y, Hori R, Komano T. (1993). P-glycoprotein-mediated transcellular transport of MDR-reversing agents. FEBS Lett, 324, 99–102.
- Tiwari R, Mohan M, Kasture S, Maxia A, Ballero M. (2009). Cardioprotective potential of myricetin in isoproterenol-induced myocardial infarction in Wistar rats. Phytother Res, 23, 1361–1366.
- Václavíková R, Horský S, Simek P, Gut I. (2003). Paclitaxel metabolism in rat and human liver microsomes is inhibited by phenolic antioxidants. Naunyn Schmiedebergs Arch Pharmacol, 368, 200–209.
- von Möllendorff E, Abshagen U, Akpan W, Neugebauer G, Schröter E. (1986). Clinical pharmacologic investigations with carvedilol, a new beta-blocker with direct vasodilator activity. Clin Pharmacol Ther, 39, 677–682.
- von Moltke LL, Weemhoff JL, Bedir E, Khan IA, Harmatz JS, Goldman P, Greenblatt DJ. (2004). Inhibition of human cytochromes P450 by components of Ginkgo biloba. J Pharm Pharmacol, 56, 1039–1044.
- Xue B, Li J, Chai Q, Liu Z, Chen L. (2008). Effect of total flavonoid fraction of Astragalus complanatus R. Brown on angiotensin II-induced portal-vein contraction in hypertensive rats. Phytomedicine, 15, 759–762.
- Zarghi A, Foroutan SM, Shafaati A, Khoddam A. (2007). Quantification of carvedilol in human plasma by liquid chromatography using fluorescence detection: application in pharmacokinetic studies. J Pharm Biomed Anal, 44, 250–253.