Abstract
Context: The recent developments in non-viral gene therapy and DNA vaccine have fostered the development of efficient plasmid DNA (pDNA) purification processes.
Objectives: This work aimed to establish a cost-effective purification process for the large-scale production of plasmid DNA for gene therapy and DNA vaccine.
Materials and methods: E. coli DH5α harboring pCDNA3.1-GFP (7200 base pairs) was used as a model plasmid. Hydrophobic-interaction chromatography (HIC) was employed to purify supercoiled plasmid DNA (sc pDNA).
Results: With this method, not only host contaminants, but also open circular plasmid DNA (oc pDNA) could be removed from sc pDNA. Anion-exchange HPLC analysis proved that the recovery of HIC could reach 75%. The plasmid DNA exhibited high purity with supercoiled percentage of 98 ± 1.2% and undetectable residual endotoxins, genomic DNA, RNA and protein. The purity of pDNA had nothing to do with the flow rate in the range at least up to 400 cm/h. Liposomes transfection experiment prove that the purified pDNA in this article had higher transfection efficiency than the control pDNA.
Discussion and conclusion: In the present work, we confirmed the possibility of separation of sc pDNA from oc pDNA and other host contaminants using a single HIC chromatography.
Keywords::
Introduction
In recent years, DNA vaccine and gene therapy have attracted a lot of interest as new ways of preventing or treating diseases through gene transfer (CitationRobinson, 1999; CitationMountain, 2000). DNA vaccine is safer and more efficient than classic vaccines such as life-attenuated viruses, inactivated microorganisms, etc. DNA vaccine can encode and process the antigen in the organism, so the process is like an actual infection and antigen protein is nearly identical (different than recombinant protein vaccines) to the antigen pattern of an infection (CitationSchleef & Schmidt, 2004). As of 2011, 18.7% of all gene therapy clinical trials utilized pDNA as the vector (www.wiley.co.uk/genmed/clinical), just behind adenoviruses (24.2%) and retroviruses (20.7%). Compared with viral vectors, non-viral vectors such as pDNA is more attractive due to safety, versatility, ease of preparation and it has the benefit of not inducing immunogenic reactions against components of the vector itself upon administration to patients (CitationVoss, 2007).
Plasmids are usually produced in an Escherichia coli host by fermentation and are purified by a sequence of operations (CitationPrazeres et al., 1998). The processes of production and purification often result in the generation of other isoforms such as oc, linear and denatured pDNA (CitationSchleef & Schmidt, 2004). The resulting isoforms are less efficient than sc isoforms at inducing a complete profile of immune responses if cleavage or strand melting disrupts promoter or gene coding regions. Evidence has shown that high sc levels are required for eliciting an effective immune response and protection from infectious challenge (CitationCupillard et al., 2005; CitationPillai et al., 2008). So, it is the key step to separate sc pDNA from other isoforms in the large-scale purification of pDNA vectors for DNA vaccine and gene therapy.
Some chromatographic techniques have been used to purify pDNA, such as anion-exchange chromatography (AEC) (CitationHines et al., 1992; CitationPrazeres et al., 1998; CitationFranc et al., 2010), size-exclusion chromatography (SEC) (CitationHorn et al., 1995), hydrophobic-interaction chromatography (HIC) (CitationGurunathan et al., 2000; CitationDiogo et al., 2003; CitationDuarte et al., 2004; CitationMoreira et al., 2005; CitationPereira et al., 2010) and affinity chromatography (AC) (CitationWils et al., 1997; CitationSchluep & Cooney, 1998; CitationSandberg et al., 2004). These chromatographic methods work well in removing host contaminants (protein, RNA, endotoxins, genomic DNA, etc.), but they often use two-step or more chromatographic steps which will increase costs and decrease the recovery of pDNA. Also, they cannot remove oc pDNA from sc pDNA just by a single chromatographic step; the oc isoforms are less efficient than sc isoforms at inducing a complete profile of immune responses. Some methodology use organic reagent such as isopropanol (CitationDuarte et al., 2004), which will increase the environmental pollution and the hazards to human. In this article, we will try to purify pDNA and remove oc pDNA from sc pDNA by selecting appropriate HIC column and improving the loading method and elution method; we will try to purify sc pDNA by a single HIC column in order to decrease costs and increase the recovery of sc pDNA; we will not use the organic reagent in the whole process in order to decrease the environmental pollution. In our laboratory, we have used AEC to purify pDNA, although the purity of pDNA can fulfill the requirements of regulatory authorities, but the purity will be affected if the flow rate exceeds 120 cm/h (CitationLi et al., 2011). In this article, we will try to improve the purification efficiency by increasing the flow rate.
Materials and methods
Materials
The E. coli host strain is DH5α. DH5α was purchased from Takara. Plasmid pCDNA3.1-GFP, constructed by the Institute of Bio-Pharmacology of Guangdong Pharmaceutical University, was used as a model plasmid.
Octyl Sepharose 4 Fast Flow, Sephadex G-25, Mono QTM 5/50 GL, XK 16/40 column, AKTA Basic system and Frac-920 fraction collector were all purchased from Amersham Bioscience. Beckman Avanti J-25 centrifuge was provided by Beckman Co. Tianli gel scanner was purchased from Tianli Biotech.
Fetal bovine serum (FBS) was from TBD, Lipofectamine 2000 and RPMI 1640 were from Invitrogen, agarose was from FMC and Luria broth (LB) was from Sigma, and Plasmid mini kit was from OMEGA.
Other reagents used were of analytical-reagent grade or with equivalent purity.
Bacterial culture
E. coli DH5α harboring pCDNA3.1-GFP was inoculated into a 2000 mL flask containing 500 mL of LB medium supplemented with 100 µg/mL Ampicillin and incubated overnight at 37°C and 200 rpm in a rotary shaker. Bacteria were harvested at late log phase. And 1 mL of culture was used to purify pDNA using OMEGA plasmid kit as control.
Lysis and primary isolation
Bacteria were lysed using a modification of the alkaline method described by CitationBirnboim and Doly (1979). Cells were harvested and centrifuged at 5000g for 15 min at 4°C. The supernatants were discarded and the bacterial pellets were resuspended in 25 mL of 25 mM Tris, 50 mM glucose, and 10 mM EDTA (pH 8.0). Alkaline lysis was performed by adding an equal volume of prechilled 200 mM NaOH and 1% (w/v) sodium dodecylsulfate solution (SDS) on ice for 3 min. The lysate was neutralized with 20 mL of prechilled (on ice) 3 M potassium acetate (pH 5.0) for 5 min. The precipitate was removed by centrifugation at 12,000g for 20 min at 4°C. Next, the neutralized lysate was in triplicate, and solid ammonium sulfate was dissolved in the neutralized lysate up to concentrations 2 M, 2.5 M and 3 M, respectively. Samples were agitated to dissolve the salt and subsequently incubated for 15 min at 4°C, and then centrifuged at 12,000g for 20 min at 4°C. The supernatant was filtered through 0.45 um filter, and then loaded directly on the HIC columns.
Hydrophobic-interaction chromatography
XK 16/40 (40 × 1.6 cm) column was packed with 40 mL of Octyl Sepharose 4 Fast Flow adsorbent according to the manufacturer’s instructions. Chromatography was performed with AKTA Purifier system. Three different loading buffers were prepared: (i) 2 M ammonium sulfate in 40 mM Tris/HCl, pH 8.0; (ii) 2.5 M ammonium sulfate in 40 mM Tris/HCl, pH 8.0; (iii) 3 M ammonium sulfate in 40 mM Tris/HCl, pH 8.0. The salt concentration of loading buffer is in accord with the plasmid sample. Elution buffer was always 1 M ammonium sulfate in 40 mM Tris/HCl (pH 8.0). The flow rate was 400 cm/h. Absorbance was monitored at 260 nm.
The column was equilibrated with 2 column volumes (CV) of loading buffer. Plasmid samples were then injected on column. The column was washed with 2 CV of loading buffer. A 10 CV linear gradient of loading buffer to elution buffer was applied to resolve contaminants and pDNA. Plasmid-containing fractions were collected and analyzed for contaminants and plasmid.
The column was regenerated with 4 CV of water, sanitized with 2 CV of 1 M NaOH followed by re-equilibration with loading buffer.
Desalting
Plasmid samples were desalted prior to all of the analysis steps to prevent the interference of the salt on the analysis. Sephadex G-25 column was used with 40 mM Tris/HCl (pH 8.0) as running buffer.
Agarose gel electrophoresis
Samples from HIC were analyzed by horizontal electrophoresis in 0.8% agarose gels in TAE buffer (40 mM Tris base, 20 mM acetic acid and 1 mM EDTA, pH 8.0). Electrophoresis was run at 100 V. Gels were stained with 0.5 µg/mL ethidium bromide and scanned by the Tianli image system. The homogeneity of pDNA was analyzed by Tianli gel analysis software. The homogeneity was expressed as the percentage of the sc pDNA in total pDNA.
Analytical chromatography
Plasmid purity was determined by anion-exchange HPLC on a MONO QTM 5/50 GL column. Chromatography was performed with AKTA Purifier system. The column was equilibrated with 2 CV of loading buffer (40 mM Tris/HCl, pH 8.0) at a flow rate of 0.5 mL/min. Injection of 100 µL of sample was followed by a linear gradient of 0–1 M NaCl in 40 mM Tris/HCl, pH 8.0 for 10 CV. The column was cleaned with 2 CV of 1 M NaOH.
Spectrophotometry analysis
Plasmid purity was detected with spectrophotometer (Thermo). Purity was expressed as the ratio of absorbance at 260 nm and at 280 nm, respectively (A260/A280).
Protein analysis
Protein concentration was estimated using the bicinichoninic acid (BCA) assay from Bioteke (China). Samples (20 µL) were added into 200 µL BCA working reagent in a microplate. After homogenization, the microplate was incubated at 37°C for 30 min. Absorbance was measured at 570 nm in a microplate reader. A calibration curve was made with bovine serum albumin (BSA).
Endotoxin analysis
Analysis for endotoxin contamination was performed using the kinetic-QCL Limulus amebocyte lysate (LAL) assay kit from Biowhittaker (Walkersville, MD). The detection level of the method was 0.010 EU/mL.
Biological activity detection
The biological activity of plasmid was detected by a liposomes transfection experiment. 7402 cell lines were used for transfection experiment. Transfection was performed using Lipofectamine 2000 (Invitrogen, USA) according to the manufacture’s instructions. The concentration of plasmid was adjusted to 0.02 µg/µL. Transfection efficiency was measured by inverted fluorescence microscope (Olympus IX71) and flow cytometry (Beckman coulter) at 24 h after transfection.
Results
Hydrophobic-interaction chromatography
Plasmid pCDNA3.1-GFP was used as model plasmid. Cells were lysed and clarified by conventional “alkaline lysis procedures”. In order to further remove impurities from the clarified lysates and prepare sample for further purification, ammonium sulfate precipitation step was taken. Agarose gel electrophoresis analysis showed that most RNA could be removed by the ammonium sulfate precipitation step.
Next, HIC was used to remove host contaminants such as protein, RNA, endotoxin and oc pDNA from sc pDNA. To determine the effect of salt concentration on the performance of HIC column, the column was run in three different loading buffers. When the salt concentration was 2 M, only two elution peaks were obtained (). However when the salt concentration of loading buffer and plasmid sample were 2.5 M and 3 M, similar chromatograms were obtained ( and ). The chromatograms include a large flow-through peak and three elution peaks. A small but clear peak (first elution peak) was followed by a large sharp peak (second elution peak), and the third elution peak is a large wide peak. Compared to 2 M, the first small clear elution peak appeared when the salt concentration was 2.5 M and 3 M. The results from agarose gel electrophoresis showed that the flow-through peak (2 M, 2.5 M and 3 M), the first small clear elution peak (only 2.5 M and 3 M), the second large sharp elution peak (2 M, 2.5 M and 3 M) and the last elution peak (2. 2.5, and 3 M) corresponded to non-DNA impurities, oc pDNA, sc pDNA and RNA, respectively ().
Figure 1. Chromatographic separation of pDNA isoforms with HIC. Linear gradient was performed at 400 cm/h by decreasing the ammonium sulfate concentration. (A) 2–1 M ammonium sulfate in 40 mM Tris/HCl, pH 8.0, (B) 2.5–1 M ammonium sulfate in 40 mM Tris/HCl, pH 8.0, (C) 3–1 M ammonium sulfate in 40 mM Tris/HCl, pH 8.0.
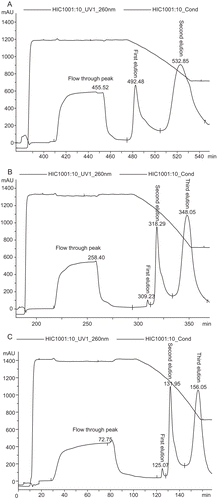
Figure 2. Agarose gel electrophoresis analysis. Lane 1: lysate; Lane 2, 6, 12: loading sample; Lane 3, 7, 13: Flow-through peak; Lane 4, 8, 14: First elution peak; Lane 5, 9, 15: Second elution peak; Lane 10, 16: Third elution peak; Lane 17: DNA ladder.
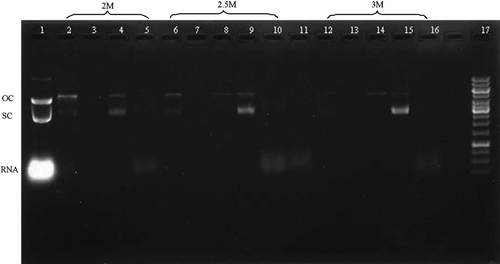
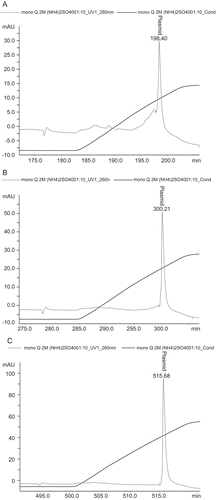
The quality of the purified pDNA was estimated by a series of analytics. Agarose gel electrophoresis () revealed that genomic DNA and RNA were absent from the plasmid product (the second large sharp elution peak) no matter what methods were used, however oc pDNA could be removed from sc pDNA only when the salt concentration were 2.5 and 3 M. The plasmid DNA exhibited high purity with supercoiled percentage of 98 ± 1.2% when the salt concentration was 3 M. However, the homogeneity was 91 ± 1.4 and 77 ± 1.7% when the salt concentrations were 2.5 and 2 M respectively. HPLC analysis proved that impurities were not detected when the salt concentration was 3 M (). The protein contaminants were not detected by the BCA method, and endotoxin analysis revealed that the level of endotoxins in the pDNA product was lower than 0.003 EU/µg pDNA, which well complied with the specification of <0.1 EU/µg pDNA for gene therapy. The A260/A280 ratio of the final pDNA is 1.91 ± 0.05 when the salt concentration was 3 M, which was also regarded as high purity.
Figure 3. Plasmid purity analysis by HPLC. Plasmid purity was determined by anion-exchange HPLC on a MONO QTM 5/50 GL column. A linear gradient was performed at 0.5 mL/min by increasing the NaCl 0–1 M in 40 mM Tris/HCl, pH 8.0 for 10 CV at a flow rate of 0.5 mL/min. (A) plasmid sample purified by 2 M ammonium sulfate, (B) plasmid sample purified by 2.5 M ammonium sulfate, (C) plasmid sample purified by 3 M ammonium sulfate.
Based on the above analysis, we have purified different size plasmids in the presence of 3 M ammonium sulphate in our laboratory. The recovery of sc pDNA was about 75%.
Biological activity detection
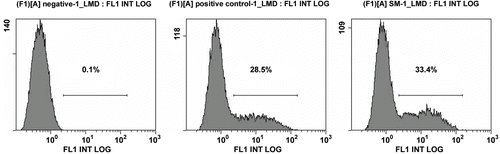
By in vitro transfection experiments, we found that the purified pDNA in this article (used 3 M) had higher transfection efficiency than the control pDNA, although the OMEGA kit was widely used to prepare pDNA for gene therapy throughout the world. By inverted fluorescence microscope analysis, we found that green fluorescent protein expressed in most cells in the experimental group (). Next, we used flow cytometry to further measure the accurate transfection efficiency. The result showed that the transfection efficiency of pDNA sample purified by 3 M was 33.6 ± 0.7%, however that of control pDNA was 28.4 ± 0.5% ().
Figure 4. Result of inverted fluorescence microscope (×100). A: Plasmid purified using OMEGA kit B: Plasmid purified by 3 M ammonium sulfate.
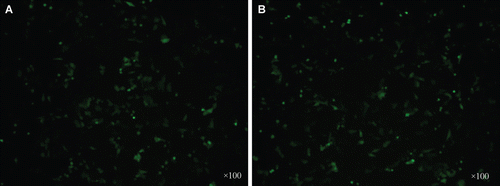
Figure 5. Transfection efficiency detected by flow cytometry. (A) negative (B) Plasmid purified using OMEGA kit C: Plasmid purified by 3 M ammonium sulfate.
Discussion
This work shows that purification processes based on a single HIC column can be used not only to remove host contaminants such as protein, RNA, endotoxin but also to remove oc pDNA from sc pDNA. In the presence of 3 M ammonium sulphate, the sc pDNA are more hydrophobic than oc pDNA. So the oc pDNA was eluted firstly from the column. Under these conditions, some oc pDNA can be removed from sc pDNA. Various evidence has shown that high supercoiled levels are required for eliciting an effective immune response and protection from infectious challenge (CitationCupillard et al., 2005; CitationPillai et al., 2008). By in vitro transfection experiments, we further prove that the final pDNA purified in present work is more effective in eliciting an effective immune response and protection from infectious challenge.
Chromatography is easy to scale up and has higher resolution. So, it is often used to produce pDNA for DNA vaccine and gene therapy. To remove trace contaminants and separate sc pDNA from oc pDNA, multiple chromatographic steps are frequently used. But the more chromatographic steps are used, the more money is spent. The recovery of pDNA is decreased with more chromatographic steps. In this work, we used one single HIC column to purify pDNA. So, the method presented in this study is more cost-effective, scalable and easier to control quality.
For the production of pharmaceutical-grade pDNA at an industrial scale, three main requirements have to be met. First, the product must be free from bacterial genomic DNA, RNA, protein and endotoxins. Second, the pDNA product must be of high homogeneity, meaning most plasmid is the supercoiled isoform. Last, the process must be cost-effective, scalable and easy to control quality.
Quality analysis (agarose gel electrophoresis, HPLC, etc.) of the final plasmid shows that the final pDNA exhibits high purity with a supercoiled percentage of 98 ± 1.2% and undetectable residual endotoxins, genomic DNA, RNA and protein. A liposome transfection experiment proves the purified pDNA described here has higher transfection efficiency than the control pDNA. So the method presented in this study meets the main requirements for the production of pharmaceutical-grade pDNA. It is easy to transfer from lab-scale preparation to large-scale production.
Conclusions
This work demonstrates that it is possible to use a single HIC chromatography to purify sc pDNA from other isoforms and host contaminants. The process is cost-effective, scalable and easy to control quality.
Declaration of interest
This work was supported by National Scientific and Technological Major Special Project for “Significant Creation of New Drugs” (No. 2009ZX09103-708); and National Natural Science Foundation of China (No. 31100664); and Natural Science Foundation of Guangdong Province (No. 10151022401000024).
References
- Birnboim HC, Doly J. (1979). A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res, 7, 1513–1523.
- Cupillard L, Juillard V, Latour S, Colombet G, Cachet N, Richard S, Blanchard S, Fischer L. (2005). Impact of plasmid supercoiling on the efficacy of a rabies DNA vaccine to protect cats. Vaccine, 23, 1910–1916.
- Diogo MM, Queiroz JA, Prazeres DM. (2003). Assessment of purity and quantification of plasmid DNA in process solutions using high-performance hydrophobic interaction chromatography. J Chromatogr A, 998, 109–117.
- Duarte M, Maria M, Joao A. (2004). Purification of plasmid DNA by hydrophobic interaction chromatography. US Patent Application 2004/0038393.
- Franc S, Ales P, Mateja C, Sandra K, Peter R, Ales S, Matjaz P. (2010). Preparation of pharmaceutical-grade plasmid DNA using methacrylate monolithic columns. Vaccine, 28, 2039–2045.
- Gurunathan S, Klinman DM, Seder RA. (2000). DNA vaccines: Immunology, application, and optimization. Annu Rev Immunol, 18, 927–974.
- Hines RN, O’Connor KC, Vella G, Warren W. (1992). Large-scale purification of plasmid DNA by anion-exchange high-performance liquid chromatography. BioTechniques, 12, 430–434.
- Horn NA, Meek JA, Budahazi G, Marquet M. (1995). Cancer gene therapy using plasmid DNA: Purification of DNA for human clinical trials. Hum Gene Ther, 6, 565–573.
- Li H, Bo H, Wang J, Shao H, Huang S. (2011). Separation of supercoiled from open circular forms of plasmid DNA, and biological activity detection. Cytotechnology, 63, 7–12.
- Moreira KA, Diogo M, Prazeres DM, Filho JLL, Porto ALF. (2005). Purification of plasmid (pVaxLacZ) by hydrophobic interaction chromatography. Braz Arch Boil Technol, 48, 113–117.
- Mountain A. (2000). Gene therapy: The first decade. Trends Biotechnol, 18, 119–128.
- Pereira LR, Prazeres DM, Mateus M. (2010). Hydrophobic interaction membrane chromatography for plasmid DNA purification: Design and optimization. J Sep Sci, 33, 1175–1184.
- Pillai VB, Hellerstein M, Yu T, Amara RR, Robinson HL. (2008). Comparative studies on in vitro expression and in vivo immunogenicity of supercoiled and open circular forms of plasmid DNA vaccines. Vaccine, 26, 1136–1141.
- Prazeres DM, Schluep T, Cooney C. (1998). Preparative purification of supercoiled plasmid DNA using anion-exchange chromatography. J Chromatogr A, 806, 31–45.
- Robinson HL. (1999). DNA vaccines: Basic mechanism and immune responses (Review). Int J Mol Med, 4, 549–555.
- Sandberg LM, Bjurling A, Busson P, Vasi J, Lemmens R. (2004). Thiophilic interaction chromatography for supercoiled plasmid DNA purification. J Biotechnol, 109, 193–199.
- Schleef M, Schmidt T. (2004). Animal-free production of ccc-supercoiled plasmids for research and clinical applications. J Gene Med, 6 Suppl 1, S45–S53.
- Schluep T, Cooney CL. (1998). Purification of plasmids by triplex affinity interaction. Nucleic Acids Res, 26, 4524–4528.
- Voss C. (2007). Production of plasmid DNA for pharmaceutical use. Biotechnol Annu Rev, 13, 201–222.
- Wils P, Escriou V, Warnery A, Lacroix F, Lagneaux D, Ollivier M, Crouzet J, Mayaux JF, Scherman D. (1997). Efficient purification of plasmid DNA for gene transfer using triple-helix affinity chromatography. Gene Ther, 4, 323–330.