Abstract
Context: N’-(7-Fluoro-5-N-methyl-10H-indolo[3,2-b]quinolin-5-ium)-N,N-dimethylpropane-1,3-diamine iodide (SYUIQ-FM05) is a semi-synthetic derivative of cryptolepine which is from Cryptolepis sanguinolenta (Lindl.) Schlechter (Periplocaeae). This ligand inhibits telomerase activity by stabilizing the G-quadruplex structure and induces growth arrest in cancer cells.
Objective: The anticancer activity of SYUIQ-FM05 via inhibiting c-kit transcription was investigated in leukemic cells.
Materials and methods: The cytotoxicity of SYUIQ-FM05 in K562 cells was evaluated using a cell viability assay and flow cytometry (FCM) at 0.4, 2.0, 10.0 and 20.0 nM. Under the same concentrations of SYUIQ-FM05 or 100 nM imatinib mesylate (IM), quantitative polymerase chain reaction (Q-PCR) investigated transcription of c-kit and bcl-2, and western blotting analyzed the expression levels of c-Kit, total mitogen-activated protein kinase kinases (MEKs), phospho-MEK (p-MEK), total extracellular regulated protein kinases (ERKs), phospho-ERK (p-ERK), Bcl-2 and Bax.
Results: SYUIQ-FM05 inhibited cellular growth with an IC50 of 10.83 ± 0.05 nM in K562 cells. c-Kit transcription was suppressed 2.69-, 4.39-, 7.71- and 10.52-fold at 0.4, 2.0, 10.0 and 20.0 nM SYUIQ-FM05, respectively, which produced proportional loss of total c-Kit protein except IM. Both SYUIQ-FM05 and IM downregulated p-MEK and p-ERK. Furthermore, bcl-2 transcription was suppressed 1.58- and 1.86-fold at 10.0 and 20.0 nM SYUIQ-FM05, respectively, but 0.4 and 2.0 nM SYUIQ-FM05 had no effect. A decrease in Bcl-2 and an increase in Bax appeared in these treated cells.
Discussion and conclusion: These findings demonstrate that SYUIQ-FM05 could induce apoptosis in a leukemic cell line through inhibiting c-kit transcription, which supports the anticancer potency of SYUIQ-FM05 in c-Kit-positive leukemic cells.
Introduction
Cryptolepine (), the nature product from Cryptolepis sanguinolenta (Lindl.) Schlechter (Periplocaeae), belongs to the indoloquinoline family, a relatively rare group of alkaloids (CitationHolt & Petrow, 1947). It has been used as antimalarial drug since its specific structure could interact with DNA through intercalation (CitationBierer et al., 1998; CitationYang et al., 1999). Based on its structure, we have synthesized 5-N-methyl quindoline derivatives. These derivatives possess a positive charge at the 5-N position of the quindoline, which contributes to their high affinity for telomeric G-quadruplex structures. These ligands, including N′-(7-fluoro-5-N-methyl-10H-indolo[3,2-b]quinolin-5-ium)-N,N-dimethylpropane-1,3-diamine iodide (SYUIQ-FM05, ), induce growth arrest in HL60 and CA46 cells through an inhibition of telomerase activity (CitationLu et al., 2008). N-(3-(Dimethylamino)propyl)-7-fluoro-10H-indolo[3,2-b]quinolin-11-amine (SYUIQ-F05, ), another quindoline derivative, down-regulates the expression of the proto-oncogene, c-myc, by stabilizing the nuclear hypersensitivity element III1 upstream of the c-myc promoter in Hep G2 hepatocellular carcinoma and Ramos and CA46 Burkitt’s lymphoma cell lines (CitationOu et al., 2007). SYUIQ-FM05 is a 5-N-methyl quindoline derivative that exerts remarkable effects on cell growth arrest and cellular senescence and inhibits telomere elongation in leukemia cells (CitationLu et al., 2008). SYUIQ-FM05 inhibits the transcription of the bcl-2 gene by stabilizing the promoter quadruplex in HL60 cells (CitationWang et al., 2010). This inhibition suggests that these quindoline derivatives exert similar effects on quadruplex structure in other genes. This ligand also inhibits cellular viability and induces the expression of senescence-associated β-galactosidase (SA-β-Gal) (CitationOu et al., 2007). These results suggest that SYUIQ-FM05 induces leukemic cell apoptosis.
Figure 1. Structures of cryptolepine and quindoline derivatives.
The c-Kit receptor regulates cellular growth and self-renewal in hematopoiesis, the development and function of mast cells, and gametogenesis. The c-Kit receptor is a 145-kDa protein that is expressed primarily in hematopoietic stem cells (HSCs) and common myeloid progenitor (CMP) cells (CitationYarden et al., 1987; CitationLennartsson et al., 2005). The ligand of this receptor is stem cell factor (SCF), which activates c-Kit autophosphorylation. The overexpression or mutation of c-kit induces hematopoitic malignancy (CitationFerrari et al., 1993; CitationWang et al., 2005). c-Kit maintains cellular self-renewal and proliferation by increasing the anti-apoptosis protein, Bcl-2, through upregulating ERK phosphorylation (CitationDomen & Weissman, 2000; CitationScholl et al., 2008). The stimulation of the c-Kit receptor with SCF activates ERK phosphorylation in X-irradiated HL60 cells and increases the resistance to apoptosis. Interestingly, low-level ERK phosphorylation induces apoptosis, but not proliferation, in primary acute myelogenous leukemia blasts (CitationLunghi et al., 2003; CitationJalal Hosseinimehr et al., 2004). MEK activity determines ERK activity because MEK phosphorylation (p-MEK) occurs prior to ERK phosphorylation, and p-MEK directly phosphorylates ERK (CitationCrews et al., 1992). This process is modulated by c-Kit, and receptor tyrosine kinase (RTKs) inhibitors, such as imatinib mesylate, block this pathway. The RAS/MEK/ERK phosphorylation cascade that is activated by c-Kit phosphorylation is closely related to cell survival. Imatinib mesylate suppresses c-Kit receptor phosphorylation and induces cancer cell apoptosis (CitationDolci et al., 2001; CitationChu et al., 2004; CitationLasater et al., 2008). RTKs are the current primary targets of cancer chemotherapies, but the emergence of drug resistance has urged the development of novel inhibitors of c-kit function (CitationAl-Obeidi & Lam, 2000; CitationGounder & Maki, 2011; CitationKee & Zalcberg, 2012). The suppression of c-kit transcription using flavopiridol induces apoptosis in gastrointestinal stromal tumor cells through the inhibition of poly(ADP-ribose) polymerase and downstream ERK phosphorylation (CitationSambol et al., 2006). This mechanism suggests an alternative therapy for c-Kit-positive cancer.
The promoter of the c-kit gene, which encodes the c-Kit tyrosine kinase receptor, exhibits a G-quadruplex structure (CitationFernando et al., 2006). We investigated the effect of SYUIQ-FM05 on c-kit gene transcription because of the role of the c-Kit receptor in the proliferation of leukemia and hematopoietic stem/progenitor cells. We hypothesized that the repression of c-kit gene transcription would decrease c-Kit receptor levels on the cellular surface, which would affect c-Kit autophosphorylation and inhibit the activation of the RAS/MEK/ERK pathway. The expression level of Bcl-2 would be subsequently altered. The overall effects may negatively impact cancer cells.
The current study is a continuation of previous work. Because SYUIQ-FM05 possesses higher bioactivity compared to other 5-N-methyl quindoline derivatives (CitationLu et al., 2008), we investigated its effects on c-kit transcription and the RAS/MEK/ERK pathway, which may be responsible for its anticancer potency in c-Kit-positive chronic myelocytic leukemia (CML) K562 cell line.
Materials and methods
Chemicals
N′-(7-Fluoro-5-N-methyl-10H-indolo[3,2-b]quinolin-5-ium)-N,N-dimethylpropane-1,3- diamine iodide (SYUIQ-FM05) was synthesized using a previously reported method (CitationLu et al., 2008) with slight modifications. The melting point was 230–232°C. SYUIQ-FM05 was prepared in 40% 1,2-propanediol (P.E.) as a stock solution. Imatinib mesylate (IM) was obtained from Novartis Pharma (Basel, Switzerland), and an IM solution was prepared in ultrapure water. DMEM (high glucose) and Iscove’s-modified Dulbecco’s medium (IMDM) were purchased from Invitrogen (Life Technologies, MD, USA).
Cell culture
K562 cells (ATCC, VA, USA) were maintained in standard IMDM supplemented with 10% (v/v) inactivated fetal bovine serum (Gibco BRL; NY, USA), 100 U/mL penicillin, and 100 µg/mL streptomycin at 37°C in a humidified 5% CO2 atmosphere. The cells were seeded in 96-well plates (1 × 105 cells/mL) or 6-well plates (1 × 107 cells/mL) and treated with various concentrations of SYUIQ-FM05 or 100 nM imatinib mesylate. The vehicle control group was maintained in a final concentration of 0.8% (v/v) 1,2-propanediol.
Cytotoxicity assay
K562 cells were seeded in 96-well plates (Corning, NY, USA) and harvested during exponential growth to evaluate the antiproliferative effect of SYUIQ-FM05. The cells were incubated with varying concentrations of SYUIQ-FM05 for 24 h followed by a 4 h incubation with 0.1 mg (20 µL of 5 mg/mL solution) MTT at 37°C. The color intensity was measured using a microplate reader (Ascent) and Ascent Software for Multiskan (Thermo Fisher Scientific, Inc., MA, USA). Cellular viability was determined relative to the absorbance of the control group. The final value is expressed as the mean percentage of surviving cells per concentration.
Assessment of apoptosis
Cellular apoptosis was evaluated in K562 cells after treatment with various concentrations of SYUIQ-FM05 for 24 h in 6-well plates (Corning, NY, USA). The cells were stained with an Annexin V apoptosis KIT (Bestbio, Shanghai, China) containing Annexin V-FITC and propidium iodine (PI). The cells were stained in the following groups to establish the testing parameters: FITC(−)/PI(−), FITC(−)/PI(+), FITC(+)/PI(−), FITC(+)/PI(+). SYUIQ-FM05-treated cells were harvested, washed with cold PBS, suspended in 400 µL binding buffer, and incubated with Annexin V-FITC and PI, successively. The Annexin V-positive population was analyzed using a FacsCalibur flow cytometer (Becton-Dickinson, CA, USA).
Real-time fluorescent quantitative PCR
RNA isolation was performed using an RNAprep pure Cell/Bacteria Kit (Tiangen, Beijing, China) from 107 cells. Reverse transcription was performed using 2 µg RNA in a9 µL reaction system using random nine primers (Takara, Kyoto, Japan) and M-MLV reverse transcription reagents (Promega, WI, USA) in a Biometra T-personal thermocycler (Biometra, Göttingen, Germany). All real-time fluorescent quantitative PCR (qPCR) assays were performed in a ROCHE LightCycler 2.0 (ROCHE, Basel, Switzerland). The relative expression of the c-kit gene was calculated by normalizing c-kit gene expression to GAPDH. Reverse transcription was performed at 25°C for 10 min, 42°C for 60 min, and 99°C for 5 min. The sense primers for c-kit, bcl-2 and GAPDH for PCR are shown in .
Table 1. Primer sequences for PCR amplification and conditions.
Western blot
SYUIQ-FM05-treated cells were harvested, washed in cold PBS, and lysed in RIPA buffer containing 50 mM HEPES-KOH (pH 7.5), 150 mM NaCl, 1 mM EDTA, 2.5 mM EGTA, 1 mM DTT, 0.1% Tween 20, 10% glycerol, 10 mM β-glycerophosphate, 1 mM/L NaF, 0.1 mM/L Na3VO4, 0.2 mM phenylmethylsulfonyl fluoride, and PhosSTOP phophatase inhibitor tabs (ROCHE, Basel, Switzerland). Cell lysates containing 80 µg protein were resolved using 10% SDS-PAGE, transferred to Immuno-Blot polyvinylidene difluoride membranes (Millipore, MA, USA) and probed with antibodies, including KIT, α-tubulin (Cell Signaling Technology, MA, USA), p-MEK, total MEK, p-ERK, total ERK, Bcl-2, and Bax (Santa Cruz Biotechnology, CA, USA). Detection was performed using enhanced chemiluminescence reagents (Tiangen, Beijing, China). The membrane was stripped prior to total MEK and total ERK detection and incubated with the corresponding detective antibody. The band intensity was quantified using BIO-RAD Quantity One software.
Statistical analysis
SPSS v16.0 software (SPSS Inc., Chicago, IL, USA) was used for the statistical analysis. The values were expressed as the mean ± SE from three or four independent experiments. Statistical comparisons were made using Student’s t-test and one-way analysis of variance (ANOVA), followed by Tukey’s test. The level of significance was set at p < 0.05 or p < 0.01.
Results
Cellular toxicity of SYUIQ-FM05 in vitro
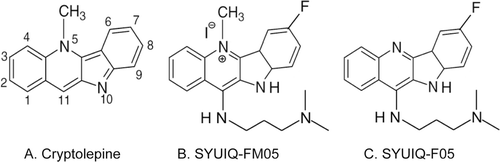
SYUIQ-FM05 inhibited the proliferation of K562 cells in the MTT assay in a concentration- and time-dependent manner. The IC50 of SYUIQ-FM05 for cellular proliferation was 10.83 ± 0.05 nM. Cellular growth was obviously suppressed at 2.0, 10.0 and 20.0 nM SYUIQ-FM05 (p < 0.05). The maximal effect of 67.3% inhibition was observed with a 24 h incubation of 20.0 nM SYUIQ-FM05 (), which was approximately 3.38-fold greater than the inhibition in the vehicle control (19.7%). No obvious difference between the vehicle control and 0.4 nM SYUIQ-FM05 was observed. The relative proliferation of K562 cells was markedly reduced by 71.6, 69.5, 64.3 and 29.0% with 3, 6, 12 and 24 h exposures to SYUIQ-FM05, respectively (). Significant differences between SYUIQ-FM05 concentrations were observed at each time point with a similar trend of inhibitory effects.
Figure 2. Cytotoxicity profile of SYUIQ-FM05-treated cells. (A) Exponentially growing K562 cells were treated with various concentrations of SYUIQ-FM05 and cell viability was assessed after 24 h. (B) Dose- and time-dependent cytotoxicity profile of K562 cells following SYUIQ-FM05 exposure. Cell viability was evaluated at four time points, and the cell viability ratio was normalized to vehicle controls. Experiments were performed in 96-well plates and assessed using the MTT assay. The data shown are representative of the combined means ± SE from three independent experiments. *p < 0.05 versus vehicle control.
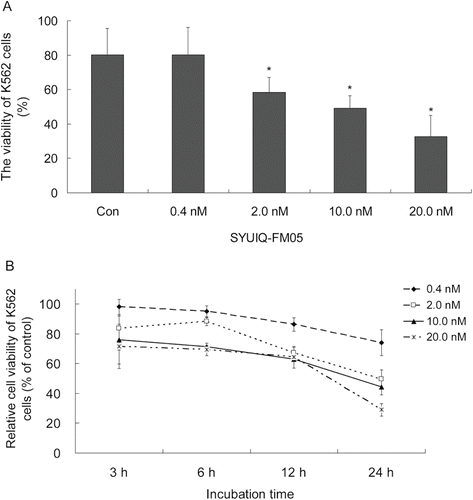
We evaluated the apoptotic response using Annexin V-FITC/PI labeling to characterize the cytotoxicity profile of SYUIQ-FM05 in K562 cells. As shown in , 0.4 nM SYUIQ-FM05 produced no obvious apoptotic response (ratio of 10.53%), which was similar to vehicle control (11.65%), after a 24 h exposure. However, a gradual increase in cellular apoptosis (22.05, 31.82 and 32.92%) was observed after exposure to 2.0, 10.0 and 20.0 nM SYUIQ-FM05, respectively (p < 0.05). The inhibition of cellular apoptosis by SYUIQ-FM05 was concentration-dependent.
Figure 3. SYUIQ-FM05-induced apoptosis in K562 cells. K562 cells were treated with 0.4, 2.0, 10.0 and 20.0 nM SYUIQ-FM05 for 24 h and harvested for the apoptosis assay. Cell apoptosis was detected using Annexin V-FITC/PI double-staining flow cytometry. The figures are representative flow cytometric dot plots of the cell population with Annexin-V FITC staining (X-axis), PI-staining (Y-axis), or both stains. Four cell populations are shown: normal cells (Annexin V-FITC/PI negative, lower left quadrant), early apoptotic cells (Annexin V-FITC positive/PI negative, lower right quadrant), late apoptotic cells and necrotic cells (Annexin V-FITC /PI positive, upper right quadrant), and injured cells (Annexin V-FITC negative/PI positive, upper left quadrant). The numbers represent the percentage of the cell population in each of the respective gates. The dot plots are representative of one experiment. These experiments were repeated four times.
SYUIQ-FM05-induced downregulation of c-kit transcription reduced total c-Kit in K562 cells
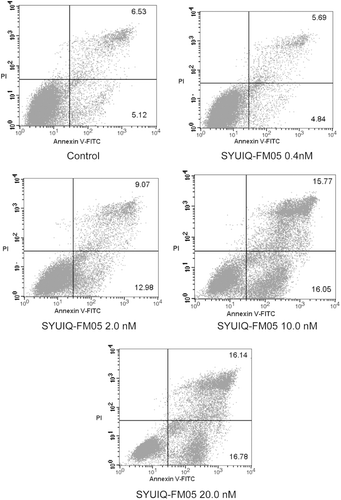
Real-time quantitative PCR was performed to determine the relative expression of c-kit mRNA and investigate the effect of SYUIQ-FM05 on c-kit gene transcription. The exposure of K562 cells to SYUIQ-FM05 for 24 h robustly inhibited c-kit gene transcription at different concentrations (). SYUIQ-FM05 at 0.4, 2.0, 10.0 and 20.0 nM suppressed c-kit mRNA expression by 2.69-, 4.39-, 7.7- and 10.52-fold (p < 0.01), respectively, compared to the vehicle control. These data demonstrated a concentration-dependent inhibition of c-kit transcription, which corresponded to the antiproliferative and pro-apoptotic effects.
Figure 4. SYUIQ-FM05 reduced the mRNA expression of the proto-oncogene c-kit. K562 cells were treated with various concentrations of SYUIQ-FM05 or 100 nM IM, and the expression of c-kit mRNA was determined using real-time fluorescent quantitative RT-PCR. Columns indicate the relative c-kit expression normalized to GAPDH mRNA; bars are the mean standard deviation. The data are representative of the combined means ± SE from four independent experiments. *p < 0.01, #p < 0.01 and **p < 0.001 versus vehicle control.
The expression of total c-Kit on Western blots also revealed a suppressive effect of SYUIQ-FM05 in K562 cells (). The maximal inhibitory effect was observed with 20.0 nM SYUIQ-FM05 for 24 h (p < 0.05). SYUIQ-FM05-treated cells (0.4, 2.0 and 10.0 nM) also exhibited obvious losses in total c-Kit (p < 0.05). Imatinib mesylate (IM) was chosen as a positive control in follow-up studies because this compound did not alter total c-Kit expression.
Figure 5. Effect of SYUIQ-FM05 on the expression of total c-Kit, p-MEK, total MEK, p-ERK, total ERK, Bcl-2 and Bax. Cell lysates of SYUIQ-FM05- and 100 nM IM-treated cells were subjected to Western blot analysis at 24 h. The membranes were probed for (A) c-Kit, (B) p-MEK, total MEK, p-ERK, total ERK and (C) Bcl-2 and Bax; α-tubulin confirmed equal protein loading. The blots were performed on three independent occasions with similar results, and the presented result is representative of one experiment. The quantification of band intensity is detailed in supplementary files 1 and 2, available online.
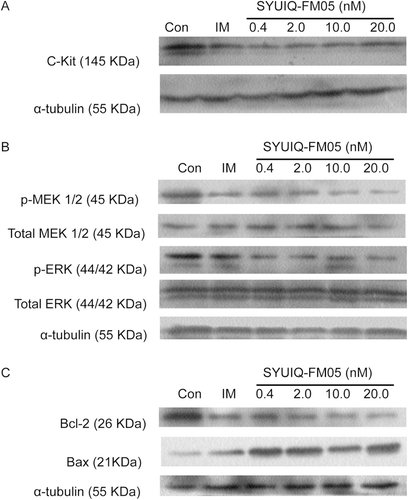
The loss of total c-Kit inhibited MEK activity and ERK phosphorylation
The loss of total c-Kit downregulated c-Kit autophosphorylation, which attenuated MEK activity via the blockade of MEK phosphorylation at elevated SYUIQ-FM05 concentrations and 100 nM IM. The inhibition of MEK phosphorylation correspondingly downregulated ERK phosphorylation. SYUIQ-FM05 exposure for 24 h significantly downregulated p-MEK and p-ERK (). p-MEK expression was inhibited significantly at 0.4, 2.0, 10.0 and 20.0 nM SYUIQ-FM05 and IM (p < 0.05) compared to the vehicle control. The 42 kDa p-ERK was rarely detected, and the downmodulation of 44 kDa p-ERK was maximal at 20.0 nM SYUIQ-FM05 (p < 0.05). Similarly, 0.4, 2.0 and 10.0 nM SYUIQ-FM05 also obviously inhibited ERK phosphorylation (p < 0.05). However, 100 nM IM also downregulated p-ERK (p < 0.05). No changes in total MEK or ERK expression were observed in SYUIQ-FM05-treated cells.
SYUIQ-FM05 downregulated bcl-2 transcription and increased Bax expression
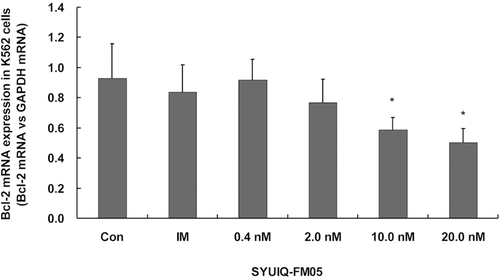
The level of bcl-2 transcription and Bcl-2 expression were determined using Q-PCR and Western blot analysis, respectively, to confirm the effect of SYUIQ-FM05 on apoptosis. SYUIQ-FM05 appreciably inhibited bcl-2 transcription at 10.0 and 20.0 nM (p < 0.05) (). SYUIQ-FM05 did not obviously decrease bcl-2 transcription at 0.4 or 2.0 nM. IM did not alter bcl-2 transcription. An obvious reduction in Bcl-2 expression was observed in all SYUIQ-FM05- and IM-treated cells (). Maximal suppression of Bcl-2 expression was observed at 10.0 and 20.0 nM SYUIQ-FM05 (p < 0.01). Moderate inhibition of Bcl-2 expression was observed with 0.4 and 2.0 nM SYUIQ-FM05 (p < 0.05). Bax expression was gradually upregulated with increasing SYUIQ-FM05 concentrations (p < 0.05) and IM (p < 0.01). These data supported the definite inhibitory effect of SYUIQ-FM05 on Bcl-2 expression and Bax upregulation.
Figure 6. Effect of SYUIQ-FM05 on bcl-2 transcription. K562 cells were treated with various concentrations of SYUIQ-FM05 or 100 nM IM, and the expression of bcl-2 mRNA was determined using real-time fluorescent quantitative RT-PCR. The columns are the relative bcl-2 expression normalized to GAPDH mRNA; bars are the standard deviation. The data are representative of the combined means ± SE from four independent experiments. *p < 0.05 versus vehicle control.
Discussion
The present study is the first demonstration that a 5-N-methyl quindoline derivative, SYUIQ-FM05, downregulated the transcription of pro-oncogene c-kit and significantly diminished total c-Kit. An absolute decrease in c-Kit may absolutely decrease phosphorylated c-Kit (CitationSambol et al., 2006), which would block the activation of downstream events that transduce proliferation signals in K562 cells. This effect of SYUIQ-FM05 was demonstrated by the down-modulation of ERK phosphorylation. The anti-apoptotic protein, Bcl-2, was down-regulated via reduced bcl-2 transcription and Bcl-2 clearance, which was induced by lower p-ERK levels. All of these factors contributed to apoptosis in leukemic cells.
We evaluated the cell growth arrest effect of SYUIQ-FM05 on the K562 cell line based on the inhibitory proliferation effect of SYUIQ-FM05 on HL60 and CA46 cells (CitationLu et al., 2008). SYUIQ-FM05 inhibited the proliferation of cells from hematological malignancy in a dose-dependent manner. We also tested the cytotoxicity of SYUIQ-FM05 in cells incubated for 24 h, and cellular proliferation was suppressed in a time-dependent manner. SYUIQ-FM05 concentrations of 0.4, 2.0, 10.0 and 20.0 nM were selected for further analysis based on the dose- and time-dependent cytotoxicity and the IC50 value.
Several concentrations of SYUIQ-FM05 inhibited c-kit gene transcription. SYUIQ-FM05 correspondingly suppressed the expression of c-Kit protein. The current study lacked sufficient data to demonstrate a direct action of SYUIQ-FM05 on the G-quadruplex structure of the c-kit gene, but the loss of total c-Kit was produced by the suppression of gene transcription or the inhibition of the RNA polymerase, which down-modulated c-Kit autophosphorylation (CitationSambol et al., 2006). Receptor tyrosine kinase inhibitors, such as imatinib mesylate, do not alter c-kit gene transcription, but IM directly inhibited c-Kit autophosphorylation, which suppresses the propagation of downstream proliferation signals (CitationKrystal et al., 2000; CitationWang et al., 2000; CitationFrost et al., 2002). These data, support the differential results of c-kit gene transcription inhibition and the loss of total c-Kit in SYUIQ-FM05-treated, but not IM-treated, cells.
MEK and ERK phosphorylation was reduced in SYUIQ-FM05- and IM-treated cells. MEK and ERK proteins play important roles in the activation of the RAS/MEK/ERK cascade, which determines cell survival, and IM inhibited MEK and ERK phosphorylation (CitationFrost et al., 2002; CitationYu et al., 2003; CitationChu et al., 2004). Low IM concentrations do not induce apoptosis after a short incubation, but a slight suppression of ERK phosphorylation is observed (CitationBeppu et al., 2004). We increased the IM concentration to 100 nM to produce an immediate down-modulation of ERK after 24 h. Our data indicated that SYUIQ-FM05 and IM suppressed c-Kit autophosphorylation-induced MEK and ERK phosphorylation in the RAS/MEK/ERK pathway. The SYUIQ-FM05-induced inhibition of c-kit transcription was more effective than the direct suppression of IM-induced c-Kit autophosphorylation and the inhibition of MEK and ERK phosphorylation.
SYUIQ-FM05 downregulated Bcl-2 expression by suppressing p-ERK expression and inhibiting bcl-2 transcription. A lower level of p-ERK removes the inhibitory effect on Bax, which is only slightly expressed under normal conditions (CitationHarada et al., 2004; CitationOrelio & Dzierzak, 2007). Our data demonstrated that the down-regulation of Bcl-2 expression corresponded to the decrease in p-MEK and p-ERK, which increased Bax expression. SYUIQ-FM05 suppresses bcl-2 transcriptional activation through a stabilization of a GC-rich sequence in its promoter (CitationTodd et al., 2007; CitationWang et al., 2010). In our study, 10.0 and 20.0 nM SYUIQ-FM05 also inhibited bcl-2 transcription, but only a slight difference in the expression of Bcl-2 protein was detected. A downregulation in Bcl-2 was observed in SYUIQ-FM05-treated cells despite the lack of an obvious suppression of bcl-2 transcription at low and moderate SYUIQ-FM05 concentrations and 100 nM IM. The SYUIQ-FM05-induced suppression of Bcl-2 expression suggests a synergetic effect on the downregulation of Bcl-2 via inhibition of transcription and protein clearance through Bax upregulation. However, the extent of these two functions has not been determined. The SYUIQ-FM05-induced loss in total c-Kit may contribute to determinate antiproliferative and apoptotic effects in leukemic cells.
Conclusions
In conclusion, SYUIQ-FM05 suppressed c-kit transcription and reduced total c-Kit protein in K562 cells, which downregulated MEK activity, ERK phosphorylation, and the RAS/MEK/ERK signaling pathway. The synergetic effect of bcl-2 transcription suppression and Bax upregulation removes the anti-apoptotic activity of Bcl-2 and induces apoptosis. These results indicated that SYUIQ-FM05 inhibited c-kit transcription in c-Kit-positive cancer cells, which suggests the potency benefit of SYUIQ-FM05 in leukemic cells.
Supplementary Material
Download PDF (131.5 KB)Acknowledgments
This authors thank Prof. Lian-Quan Gu of the School of Pharmaceutical Science, Sun Yat-sen University for advice on this work.
Declaration of interest
This work was supported by the Key Projects in the National Science and Technology Pillar Program of China (Grant No. 2008BAI55B03). The authors also appreciate the financial support provided by the Science and Technology Foundation of Guangdong province (Grant No. 2007B030704008). This article is original, and it has been written by the stated authors who are all aware of its content and approve its submission. This manuscript has not been published previously, and it is not under consideration for publication elsewhere. The article will not be published elsewhere in the same form, in any language, without the written consent of the publisher if accepted.
Related Research Data
References
- Al-Obeidi FA, Lam KS. (2000). Development of inhibitors for protein tyrosine kinases. Oncogene, 19, 5690–5701.
- Beppu K, Jaboine J, Merchant MS, Mackall CL, Thiele CJ. (2004). Effect of imatinib mesylate on neuroblastoma tumorigenesis and vascular endothelial growth factor expression. J Natl Cancer Inst, 96, 46–55.
- Bierer DE, Dubenko LG, Zhang P, Lu Q, Imbach PA, Garofalo AW, Phuan PW, Fort DM, Litvak J, Gerber RE, Sloan B, Luo J, Cooper R, Reaven GM. (1998). Antihyperglycemic activities of cryptolepine analogues: An ethnobotanical lead structure isolated from Cryptolepis sanguinolenta. J Med Chem, 41, 2754–2764.
- Chu S, Holtz M, Gupta M, Bhatia R. (2004). BCR/ABL kinase inhibition by imatinib mesylate enhances MAP kinase activity in chronic myelogenous leukemia CD34+ cells. Blood, 103, 3167–3174.
- Crews CM, Alessandrini A, Erikson RL. (1992). The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science, 258, 478–480.
- Dolci S, Pellegrini M, Di Agostino S, Geremia R, Rossi P. (2001). Signaling through extracellular signal-regulated kinase is required for spermatogonial proliferative response to stem cell factor. J Biol Chem, 276, 40225–40233.
- Domen J, Weissman IL. (2000). Hematopoietic stem cells need two signals to prevent apoptosis; BCL-2 can provide one of these, Kitl/c-Kit signaling the other. J Exp Med, 192, 1707–1718.
- Fernando H, Reszka AP, Huppert J, Ladame S, Rankin S, Venkitaraman AR, Neidle S, Balasubramanian S. (2006). A conserved quadruplex motif located in a transcription activation site of the human c-kit oncogene. Biochemistry, 45, 7854–7860.
- Ferrari S, Grande A, Zucchini P, Manfredini R, Tagliafico E, Rossi E, Temperani P, Torelli G, Emilia G, Torelli U. (1993). Overexpression of c-kit in a leukemic cell population carrying a trisomy 4 and its relationship with the proliferative capacity. Leuk Lymphoma, 9, 495–501.
- Frost MJ, Ferrao PT, Hughes TP, Ashman LK. (2002). Juxtamembrane mutant V560GKit is more sensitive to Imatinib (STI571) compared with wild-type c-kit whereas the kinase domain mutant D816VKit is resistant. Mol Cancer Ther, 1, 1115–1124.
- Gounder MM, Maki RG. (2011). Molecular basis for primary and secondary tyrosine kinase inhibitor resistance in gastrointestinal stromal tumor. Cancer Chemother Pharmacol, 67, 25–43.
- Harada H, Quearry B, Ruiz-Vela A, Korsmeyer SJ. (2004). Survival factor-induced extracellular signal-regulated kinase phosphorylates BIM, inhibiting its association with BAX and proapoptotic activity. Proc Natl Acad Sci USA, 101, 15313–15317.
- Holt SJ, Petrow V. (1947). Cabarzole, carbolines, and related compounds [Part 1. Quindoline Derivatives]. J Chem Soc, 18, 607–611.
- Jalal Hosseinimehr S, Inanami O, Hamasu T, Takahashi M, Kashiwakura I, Asanuma T, Kuwabara M. (2004). Activation of c-kit by stem cell factor induces radioresistance to apoptosis through ERK-dependent expression of survivin in HL60 cells. J Radiat Res, 45, 557–561.
- Kee D, Zalcberg JR. (2012). Current and emerging strategies for the management of imatinib-refractory advanced gastrointestinal stromal tumors. Ther Adv Med Oncol, 4, 255–270.
- Krystal GW, Honsawek S, Litz J, Buchdunger E. (2000). The selective tyrosine kinase inhibitor STI571 inhibits small cell lung cancer growth. Clin Cancer Res, 6, 3319–3326.
- Lasater EA, Bessler WK, Mead LE, Horn WE, Clapp DW, Conway SJ, Ingram DA, Li F. (2008). Nf1+/- mice have increased neointima formation via hyperactivation of a Gleevec sensitive molecular pathway. Hum Mol Genet, 17, 2336–2344.
- Lennartsson J, Jelacic T, Linnekin D, Shivakrupa R. (2005). Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cells, 23, 16–43.
- Lu YJ, Ou TM, Tan JH, Hou JQ, Shao WY, Peng D, Sun N, Wang XD, Wu WB, Bu XZ, Huang ZS, Ma DL, Wong KY, Gu LQ. (2008). 5-N-Methylated quindoline derivatives as telomeric g-quadruplex stabilizing ligands: effects of 5-N positive charge on quadruplex binding affinity and cell proliferation. J Med Chem, 51, 6381–6392.
- Lunghi P, Tabilio A, Dall’Aglio PP, Ridolo E, Carlo-Stella C, Pelicci PG, Bonati A. (2003). Downmodulation of ERK activity inhibits the proliferation and induces the apoptosis of primary acute myelogenous leukemia blasts. Leukemia, 17, 1783–1793.
- Orelio C, Dzierzak E. (2007). Bcl-2 expression and apoptosis in the regulation of hematopoietic stem cells. Leuk Lymphoma, 48, 16–24.
- Ou TM, Lu YJ, Zhang C, Huang ZS, Wang XD, Tan JH, Chen Y, Ma DL, Wong KY, Tang JC, Chan AS, Gu LQ. (2007). Stabilization of G-quadruplex DNA and down-regulation of oncogene c-myc by quindoline derivatives. J Med Chem, 50, 1465–1474.
- Sambol EB, Ambrosini G, Geha RC, Kennealey PT, Decarolis P, O’connor R, Wu YV, Motwani M, Chen JH, Schwartz GK, Singer S. (2006). Flavopiridol targets c-KIT transcription and induces apoptosis in gastrointestinal stromal tumor cells. Cancer Res, 66, 5858–5866.
- Scholl C, Gilliland DG, Fröhling S. (2008). Deregulation of signaling pathways in acute myeloid leukemia. Semin Oncol, 35, 336–345.
- Todd AK, Haider SM, Parkinson GN, Neidle S. (2007). Sequence occurrence and structural uniqueness of a G-quadruplex in the human c-kit promoter. Nucleic Acids Res, 35, 5799–5808.
- Wang WL, Healy ME, Sattler M, Verma S, Lin J, Maulik G, Stiles CD, Griffin JD, Johnson BE, Salgia R. (2000). Growth inhibition and modulation of kinase pathways of small cell lung cancer cell lines by the novel tyrosine kinase inhibitor STI 571. Oncogene, 19, 3521–3528.
- Wang XD, Ou TM, Lu YJ, Li Z, Xu Z, Xi C, Tan JH, Huang SL, An LK, Li D, Gu LQ, Huang ZS. (2010). Turning off transcription of the bcl-2 gene by stabilizing the bcl-2 promoter quadruplex with quindoline derivatives. J Med Chem, 53, 4390–4398.
- Wang YY, Zhou GB, Yin T, Chen B, Shi JY, Liang WX, Jin XL, You JH, Yang G, Shen ZX, Chen J, Xiong SM, Chen GQ, Xu F, Liu YW, Chen Z, Chen SJ. (2005). AML1-ETO and C-KIT mutation/overexpression in t(8;21) leukemia: implication in stepwise leukemogenesis and response to Gleevec. Proc Natl Acad Sci USA, 102, 1104–1109.
- Yang SW, Abdel-Kader M, Malone S, Werkhoven MC, Wisse JH, Bursuker I, Neddermann K, Fairchild C, Raventos-Suarez C, Menendez AT, Lane K, Kingston DG. (1999). Synthesis and biological evaluation of analogues of cryptolepine, an alkaloid isolated from the Suriname rainforest. J Nat Prod, 62, 976–983.
- Yarden Y, Kuang WJ, Yang-Feng T, Coussens L, Munemitsu S, Dull TJ, Chen E, Schlessinger J, Francke U, Ullrich A. (1987). Human proto-oncogene c-kit: A new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J, 6, 3341–3351.
- Yu C, Rahmani M, Almenara J, Subler M, Krystal G, Conrad D, Varticovski L, Dent P, Grant S. (2003). Histone deacetylase inhibitors promote STI571-mediated apoptosis in STI571-sensitive and -resistant Bcr/Abl+ human myeloid leukemia cells. Cancer Res, 63, 2118–2126.