Abstract
Context: Peroxynitrite (ONOO−) formation triggers oxidative/nitrative stress and contributes to exacerbated myocardial ischemia/reperfusion (MI/R) injury. Catalpol, an iridoid glycoside, abundantly found in the roots of Rehmannia glutinosa L. that is included in the family Phrymaceae in the order Lamiales, endemic to China, was found to have neuroprotective effects. However, the effect of catalpol on MI/R injury has not been identified.
Objective: This study investigated whether catalpol attenuates oxidative/nitrative stress in acute MI/R.
Materials and methods: Adult male rats were subjected to 30 min of myocardial ischemia and 3 h of reperfusion and were treated with saline, catalpol (5 mg/kg, i.p., 5 min before reperfusion) or catalpol plus wortmannin (15 µg/kg intraperitoneally injected 15 min before reperfusion).
Results: Pretreatment with catalpol significantly improved cardiac functions, reduced myocardial infarction, apoptosis and necrosis of cardiomyocytes after MI/R (all p < 0.05). Meanwhile, ONOO− formation was markedly reduced after catalpol treatment (3.01 ± 0.22 vs. 4.66 ± 0.53 pmol/mg protein in vehicle, p < 0.05). In addition, catalpol increased Akt and endothelial nitric oxide synthase phosphorylation, nitric oxide (NO) production, anti-oxidant capacity and reduced MI/R-induced inducible nitric oxide synthase expression and superoxide anion (·O2−) production in I/R hearts. PI3K inhibitor wortmannin not only blocked catalpol-induced Akt activation, but also attenuated all the beneficial effects of catalpol. Suppression of ONOO− formation by either catalpol or an ONOO− scavenger uric acid (5 mg/kg) reduced myocardial infarct size in MI/R rats.
Discussion and conclusion: In conclusion, catalpol affords cardioprotection against MI/R insult by attenuating ONOO− formation, which is attributable to increased physiological NO and decreased ·O2− production.
Introduction
Cardiovascular disease (CVD) is the leading cause of death worldwide, of which myocardial infarction (MI) is one of the most devastating consequences. A major mechanism contributing to myocardial ischemia/reperfusion (MI/R) injury is the development of myocardial oxidative stress and nitrative stress, related to the formation of reactive oxygen and reactive nitrogen species. Among the pathogenesis of MI/R-induced cardiac injury, a burst of highly reactive oxygen species (ROS) generation during the reperfusion phase, including superoxide anion (·O2−), hydrogen peroxide (H2O2), and hydroxyl radical (·OH), is an appealing one which is supported by a large foundation of experimental evidence (CitationMoens et al., 2005; CitationSaeed et al., 2005; CitationZweier & Talukder, 2006). Nitric oxide (NO) derived from vascular endothelial cells and cardiomyocytes has emerged as a fundamental modulator in regulating apoptosis (CitationKim et al., 1999). Although NO is often described as highly toxic and reactive, accumulating evidences indicate that most of the cytotoxicity attributed to NO is due to the formation of peroxynitrite (ONOO−), a strong oxidative/nitrative molecular, rather than the possible toxic effect of NO itself (CitationLevrand et al., 2006; CitationLopez et al., 1997; CitationMa et al., 1997). ONOO− produced from the interaction between NO and superoxide anion, which bursts during early reperfusion. Thus, excess or uncontrolled superoxide anion formation can shift the actions of available NO from a useful cellular signal to ONOO− and redox-related toxic products. ONOO− has been implicated a major mechanism of myocardial injury, including substantial oxidation and potential destruction of host cellular constituents, leading to the dysfunction of critical cellular processes, disruption of cell signaling pathways and the induction of cell death through both apoptosis and necrosis (Virag et al., 2003). The generation of ONOO− may be decreased by removing the substrates for its synthesis, NO and ·O2−.
Rehmannia glutinosa L., which is included in the family Phrymaceae in the order Lamiales, endemic to China, used to treat a whole host of ailments for centuries. Catalpol (), an iridiod glucoside separated from the roots of Rehmannia glutinosa, has many biological functions such as anti-inflammation, protection of liver injury, promotion of sex hormones production, and reduction of elevated blood glucose. It has been known to inhibit apoptotic death of PC12 cells induced by H2O2, and to reduce ischemia-induced neuronal death via suppression of intracellular radical production (Li et al., Citation2004a, 2008). However, to date, whether and how catalpol attenuates MI/R injury is still unidentified.
Figure 1. The structure of catalpol.
Therefore, the aims of the present study were to determine (1) whether catalpol would provide a protection against MI/R injury, and if so, (2) to investigate the involved mechanisms.
Materials and methods
Drugs
Analytical grade catalpol (purity >98%) was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China) and dissolved in physiological saline. Wortmannin was purchased from Sigma.
Experimental protocol
The experiments were performed in adherence with the National Institutes of Health Guidelines for the Use of Laboratory Animals and were approved by the Fourth Military Medical University Committee on Animal Care. Adult male Sprague-Dawley rats were fasted overnight and anesthetized by 3% pentobarbital sodium. One polyethylene catheter was inserted into the left ventricular cavity through the right carotid artery to measure left ventricular pressure (LVP). Myocardial ischemia was produced by exteriorizing the heart through a left thoracic incision and placing a 4-0 silk and making a slipknot around the left anterior descending coronary artery (LAD). After 30 min of ischemia, the slipknot was released and the myocardium was reperfused for 3 h. Rats were randomized divided into the following groups: (1) Vehicle (saline); (2) catalpol (5 mg/kg, intraperitoneally injected 5 min before reperfusion) (Li et al., Citation2004b); (3) catalpol plus wortmannin (15 µg/kg intraperitoneally injected 15 min before reperfusion). Sham-operated control rats (sham MI) underwent the same surgical procedures with the exception of LAD occlusion. At least six rats in each group were used for all the determinations after the reperfusion.
Myocardial functional assessment
MI/R function was continuously monitored before and during the entire MI/R procedure (CitationZhang et al., 2004). The arterial blood pressure and LVP were sampled and digitally processed via a hemodynamic analyzing system (RM-6200C, Chengdu Instrument Co., China). Mean arterial blood pressure (MABP), heart rate (HR), left ventricular systolic pressure (LVSP), and the instantaneous first derivation of LVP (+LVdP/dtmax and −LVdP/dtmax) were derived by computer algorithms.
Measurement of myocardial infarct size
At the end of the reperfusion, the ligature around the coronary artery was retied and 4 ml of 2% Evans blue dye (Shanghai Chemical Reagents Co., China) was injected into the left ventricular cavity. The dye was circulated and uniformly distributed except in the region of the heart previously perfused by the occluded coronary artery (ischemic region, or area-at-risk, AAR). The heart was quickly excised, and the atria, right ventricle and fatty tissues were removed from the heart. Then, frozen at −20°C, and sliced into 1 mm thick sections perpendicular to the long axis of the heart using a heart slice chamber. Slices were incubated individually using a 12-well culture plate in 1% triphenyltetrazolium chloride (TTC, Shanghai Chemical Reagents Co., China) in phosphate buffer at pH 7.4 at 37°C for 10 min, and photographed with a digital camera. Evans blue stained areas (area-not-at-risk, ANAR), TTC stained areas (red staining, ischemic but viable tissue), and TTC staining negative areas (infarcted myocardium) in each slice were determined by planimetry on a computer with SigmaScan software. The myocardial infarct size was expressed as a percentage of infarct area (INF) over total AAR (INF/AAR × 100%).
Determination of plasma creatine kinase and lactate dehydrogenase levels
Blood samples (1 ml) were drawn at the end of 3 h reperfusion. Plasma creatine kinase (CK) and lactate dehydrogenase (LDH) levels were measured spectrophotometrically (Beckman DU 640, USA) in a blinded manner. All measurements were performed in duplicate.
Terminal deoxynucleotidyl nick-end labeling (TUNEL) assay
A double-staining technique was used, i.e., TUNEL staining for apoptotic cell nuclei and 4′,6-diamino-2-phenylindole (DAPI) staining for all myocardial cell nuclei. TUNEL staining was performed by using an In Situ Cell Death Detection Kit (Roche Molecular Biochemicals) according to the protocol provided by the manufacturer. In brief, cardiomyocytes from at least four slides per block that were randomly selected were evaluated immunohistochemically to determine the number and percentage of cells exhibiting positive staining for apoptosis. The slides were covered with the mounting medium containing DAPI for detection of total nuclei. For each slide, 10 fields were randomly chosen, and a total of 100 cells per field were counted by using a defined rectangular field area (×20 objective). The index of apoptosis was determined [number of apoptotic myocytes/the total number of myocytes counted × 100%] from a total of 40 fields per heart, and the assays were performed in a blinded manner. Heart tissue samples for TUNEL analysis were obtained from the margins of noninfarcted but ischemic (peri-infarcted) areas.
Determination of myocardial caspase-3 activity
Cardiac capase-3 activity was performed by using a caspase-3 colorimetric assay kit (Chemicon, Temecula, CA, USA) following the manufacturer’s instructions. In brief, myocardial tissue was homogenized in ice-cold lysis buffer for 30 s. The homogenates were centrifuged, supernatants were collected, and protein concentrations were measured by bicinchoninic acid method. To each well of a 96-well plate, supernatant containing 200 µg of protein was loaded and incubated with 25 µg caspase-3 substrate Ac-DEVD-pNA at 37°C for 1.5 h. The optical density was measured at 405 nm with a SpectraMax-Plus microplate spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). Myocardial caspase-3 activity was calculated using a standard curve and expressed as fold increase over the mean value of sham MI/R.
Determination of Akt, eNOS, iNOS, and gp91phox expression by Western Blot
Heart tissue samples for western blot analysis were obtained from the margins of non-infarcted but ischemic (peri-infarcted) areas. Briefly, tissue samples were lysed with lysis buffer. After sonication, the lysates were centrifuged; protein quantification with the BCA protein assay kit (Pierce) was performed to ensure equal loading amount in each lane, that is, 40 μg protein. Proteins were separated by electrophoresis on SDS-PAGE and then transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA, USA). After blocking with 5% skim milk in Tris-buffered saline at room temperature for 1 h, we incubated the membrane with an antibody against Akt, phosphorylated Akt (Cell Signaling Technology, Danvers, MA), eNOS, phosphorylated eNOS, iNOS, or gp91phox (BD Bioscience Laboratories, San Jose, CA, USA) overnight at 4°C. The membrane was then washed with PBST and incubated with the corresponding secondary antibodies at room temperature for 1 h. The blots were visualized with ECL-plus reagent and were analysis with Quantity one software (Bio-rad Co.).
Quantification of superoxide, antioxidant enzymes, and malonaldialdehyde (MDA) in the heart issue
The MDA level and activities of antioxidant enzyme superoxide dismutase (SOD) in heart homogenates were determined spectrophotometrically as previously described (Zhang et al., Citation2007b). PUFA peroxides generate MDA on decomposition. In the present study, the content of MDA was estimated in the cytosolic fraction as an indicator of lipid peroxidation using a commercial colorimetric assay kit (21012, Oxis International, Portland, OR, USA). Briefly, 50 µl of the cytosolic protein fraction or standards with known concentrations was incubated with a chromogenic reagent, N-methyl-2-phenylindole, at 45°C for 1 h, and the generated chromophore after incubation was detected by spectrophotometry at 586 nm. The MDA concentration of samples was determined according to the standard curve generated in the same setting. Results were presented as MDA (pmol) normalized to milligram protein used in the assay. The enzyme activities of SOD were measured using spectrophotometric assay kits (Calbiochem, San Diego, CA, USA) according to the manufacturer’s instructions. It was assayed by the use of tetrazolium salt for detection of superoxide generated by xanthine oxidase and hypoxanthine. In brief, samples or known standards were incubated with the provided radical detector reagent followed by the addition of xanthine oxidase to initiate the reaction. After 20 min of incubation with shaking at room temperature, the absorbance at 450 nm was measured. The results of SOD activity were expressed per milligram of protein. Myocardial superoxide content was determined by lucigenin-enhanced luminescence. Superoxide production was expressed as relative light units (RLU) per second per mg vessel dry weight (RLU/mg/s).
Measurement of NOx content in cardiac tissue
Tissue samples from AAR were rinsed, homogenized in 0.9% NaCl solution (1:10, wt/vol), and centrifuged at 3000 ×g for 5 min. The pellet was discarded. NOx concentrations in the supernatant were quantified by a NO detection kit (nitrate reductase).
Quantification of tissue nitrotyrosine content
Nitrotyrosine content in the ischemia/reperfusion (I/R) cardiac tissue, a footprint of in vivo ONOO− formation and an index of nitrative stress, was determined using a nitrotyrosine ELISA kit (Cell Sciences, Canton, MA, USA) as described in our previous study (CitationLi et al., 2007).
Statistical analysis
Values are presented as means ± SEM. Data were analyzed with one-way ANOVA (GraphPad Prism software version 5.0, San Diego, CA, USA). A probability value of <0.05 was considered to be statistically significant.
Results
Cardiac function
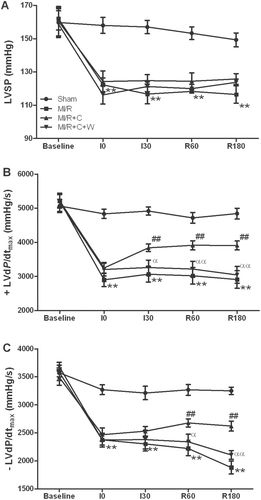
No significant differences in systemic hemodynamics were observed among all groups under baseline conditions. There were no significant differences in HR and MABP among all groups during ischemia or reperfusion period. As shown in , LVSP was significantly decreased in rats subjected to MI/R receiving vehicle only, while rats treated with catalpol markedly improved functional recovery following MI/R. In addition, treatment with catalpol significantly increased +LVdP/dtmax and −LVdP/dtmax after 3 h of reperfusion (p < 0.01). Pretreatment with wortmannin significantly blunted these beneficial effects of catalpol (p < 0.01) while wortmannin itself had no significant effect on cardiac function in rats subjected to MI/R (CitationSu et al., 2007). These results indicated that treatment with catalpol improved cardiac systolic and diastolic function in rats subjected to MI/R.
Figure 2. Effects of catalpol treatment on myocardial functions in rats subjected to 30 min of myocardial ischemia and 3 h of reperfusion (n = 8/group). LVSP: left ventricular systolic pressure; ± LVdP/dtmax: the instantaneous first derivation of left ventricle pressure; Sham: sham-operated; MI/R: myocardial ischemia/reperfusion (30 min/3 h); C: catalpol, W: wortmannin. Values presented are means ± SEM. **p < 0.01 vs. Sham, ##p < 0.01 vs. MI/R, αp < 0.05, ααp < 0.01 vs. MI/R+C.
Myocardial infarction size, plasma CK and LDH activities, and cardiomyocytes apoptosis
As shown in , 30 min of ischemia followed by 3 h of reperfusion resulted in significant myocardial infarction in the vehicle-treated rats, while no myocardial infarction was observed in the sham operated hearts. Treatment with catalpol significantly reduced the myocardial infarct size (p < 0.05). In addition, pretreatment with wortmannin before reperfusion completely abolished the protective effect of catalpol (p < 0.05).
Figure 3. Myocardial injury in rats subjected to MI/R with different treatments (n = 8/group). (A): myocardial infarct size (INF) expressed as percentage of area at risk (AAR). (B): plasma creatine kinase (CK) and lactate dehydrogenase (LDH) activities. (C): apoptotic index (percentage of TUNEL-positive nuclei in heart tissue sections). (D): myocardial caspase-3 activity. Sham: sham-operated; MI/R: myocardial ischemia/reperfusion (30 min/3 h); C: catalpol, W: wortmannin. Values presented are means ± SEM; **p < 0.01 vs. Sham, #p < 0.01, ##p < 0.01 vs. MI/R, αp < 0.05 vs. MI/R+C.
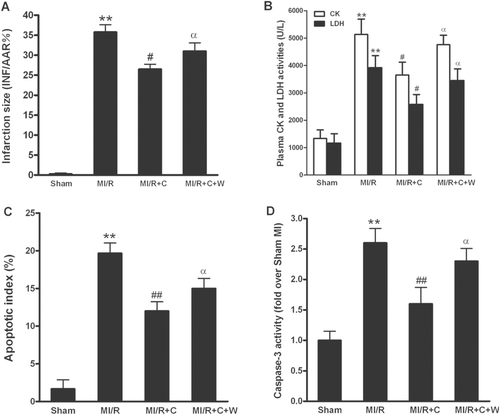
Cardiomyocyte necrosis is characterized by cell membrane disruption and cell content release. To investigate whether catalpol could decrease cardiomyocyte necrosis, plasma CK and LDH activities were measured at 3 h after reperfusion. As seen in , at 3 h after reperfusion, CK and LDH activities were markedly increased in MI/R rats. Catalpol markedly decreased the reperfusion-associated CK and LDH activities elevations compared with those in the MI/R rats (both p < 0.05). More importantly, pretreatment with wortmannin partly blunted catalpol-induced decrease of plasma CK and LDH activities (both p < 0.05 vs. catalpol group).
To further investigate whether catalpol could decrease myocardial apoptosis, TUNEL staining and caspase-3 activity in I/R cardiac tissue were determined. In myocardial tissue from the sham operated rats, a very low level of TUNEL-positive staining (AI: 1.7% ± 1.2%) was detected (). In contrast, a significant number of TUNEL-positive cells (AI: 19.7% ± 1.7%) were observed in myocardial tissue from hearts subjected to MI/R and receiving vehicle. Treatment of catalpol shortly before reperfusion exerted a significant anti-apoptotic effect, as evidenced by reduced TUNEL-positive staining (AI: 12.0% ± 1.6%, p < 0.01 vs. vehicle). Moreover, administration of wortmannin significantly blocked the catalpol-induced decrease of apoptosis (AI: 15.0% ± 1.6%, p < 0.05 vs. catalpol), while wortmannin itself at this dosage had no effects on myocardial apoptosis in MI/R rats (CitationGao et al., 2002; Zhang et al., Citation2007a). Caspase-3 is a pivotal mediator of apoptosis and myocardial caspase-3 activity is regarded as a marker of MI/R myocyte apoptosis. As shown in , MI/R also induced a significant rise in caspase-3 activity. Treatment with catalpol substantially reduced this MI/R-induced increase in myocardial caspase-3 activity (p < 0.01). Wortmannin completely abolished caspase-3 inhibitory effect of catalpol (p < 0.05). Together with the infarction size and CK/LDH levels, these data indicated that catalpol inhibited myocardial injury following MI/R in vivo, and wortmannin, a selective PI3-kinase inhibitor, blocks the protective effect of catalpol in rats subjected to MI/R.
Cardiac nitrotyrosine content
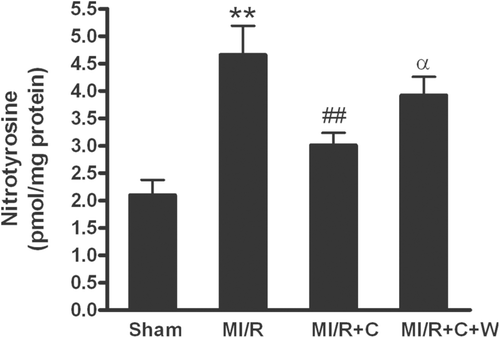
Considerable evidence demonstrated that overproduction of ONOO− and resultant oxidative/nitrative stress play a causative role in postischemic myocardial apoptosis (CitationIshida et al., 1996; CitationWang & Zweier, 1996; CitationLopez et al., 1997; CitationMa et al., 1997; CitationLevrand et al., 2006). Therefore, we examined cardiac ONOO− production. ONOO−, the biradical reaction product of ·O2− and NO, was determined by measurement of nitrotyrosine, a “footprint” of in vivo ONOO− production. As shown in , nitrotyrosine content was significantly increased in MI/R rats. Treatment with catalpol significantly reduced nitrotyrosine content (p < 0.05 vs. vehicle group). Importantly, the suppressive effect of catalpol on MI/R-induced ONOO− production was significantly blunted by PI3-kinase inhibitor wortmannin (p < 0.05 vs. catalpol group). These data suggested that catalpol exerted an antioxidative/antinitrative stress in MI/R.
Figure 4. Nitrotyrosine production in rat hearts subjected to MI/R with different treatments (n = 8/group). Sham: sham-operated; MI/R: myocardial ischemia/reperfusion (30 min/3 h); C: catalpol, W: wortmannin. Values presented are means ± SEM. **p < 0.01 vs. Sham, ##p < 0.01 vs. MI/R, αp < 0.05 vs. MI/R+C.
eNOS phosphorylation, iNOS expression and NOx content
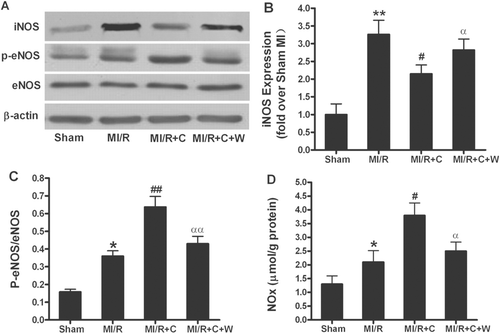
Experimental evidence demonstrating that exogenously supplied NO antagonizes the cytotoxic actions of ONOO− suggest that increased NO production may attenuate the deleterious effect of ONOO− and reduce oxidative/nitrative stress (CitationFalk et al., 2007). However, as a primary substrate for ONOO− synthesis, increased NO concentration is responsible for ONOO− overproduction. Therefore, we measured eNOS/iNOS protein expression, eNOS phosphorylation, and NO production at the end of 3 h reperfusion. Consistent with previous published results (CitationGao et al., 2002), there was no significant difference in eNOS expression among different groups; treatment with catalpol resulted in significant eNOS phosphorylation () and marked increase in NOx content (); pretreatment with wortmannin completely blocked eNOS phosphorylation and NOx increase induced by catalpol. Interestingly, in contrast to eNOS phosphorylation, the iNOS expression was increased in MI/R rats. Treatment with catalpol blocked iNOS expression in these animals, which was completely blunted by pretreatment with wortmannin ().
Figure 5. Phosphorylation of eNOS, NO production and iNOS expression by in vivo catalpol treatment and its modification by wortmannin. (A): representative blots of phosphorylated eNOS and total eNOS/iNOS in heart tissue from rats subjected to MI/R with different treatments. (B): statistical data obtained from quantitative densitometry of iNOS blots; (C): statistical data obtained from quantitative densitometry of p-eNOS/eNOS; (D): NO production in cardiac tissue. Sham: sham-operated; MI/R: myocardial ischemia/reperfusion (30 min/3 h); C: catalpol, W: wortmannin. Values presented are means ± SEM. n = 8. *p < 0.05, **p < 0.01 vs. Sham, #p < 0.05, ##p < 0.01 vs. MI/R, αp < 0.05, ααp < 0.01 vs. MI/R+C.
Cardiac oxidative stress
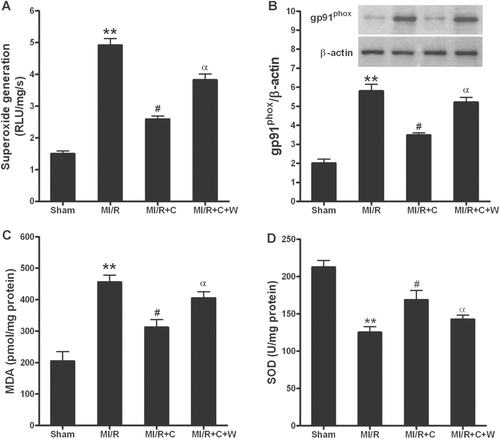
The acute reperfusion induces the enhanced generation of high ROS. Therefore, we examined cardiac superoxide generation, gp91phox protein expression, SOD activity and MDA concentration. The major source of superoxide production in the heart is NADPH oxidase. As summarized in and , MI/R-induced both superoxide generation and overexpression of gp91phox, a major component of NADPH oxidase, was completely blocked by catalpol treatment. Pretreatment with wortmannin significantly blunted this beneficial effect. The generation of MDA is widely accepted as a presumptive marker of oxidative stress. MI/R significantly increased the amount of MDA, however, catalpol treatment markedly reduced this increase in MDA formation (). In addition, antioxidant capacity was determined by the activity of SOD (·O2− scavenger) in cardiac tissue. MI/R-induced reduction in total antioxidant capacity was significantly preserved after catalpol treatment, which was also completely blunted by pretreatment with wortmannin (). These data suggested that catalpol treatment reduced cardiac oxidative stress in MI/R rats, which was significantly inhibited by wortmannin pretreatment.
Figure 6. Catalpol exerted anti-oxidative stress effects in rats subjected to MI/R. (A): superoxide production in myocardium; Myocardial superoxide content was quantified with lucigenin-enhanced luminescence; RLU, relative light units. (B): gp91phox protein expression; Inserted gel photographs are representative Western blot results from 4 to 5 animals/group. (C): malonaldialdehyde (MDA) level in myocardium. (D): activity of antioxidant enzyme superoxide dismutase (SOD) in myocardium. Sham: sham-operated; MI/R: myocardial ischemia/reperfusion (30 min/3 h); C: catalpol, W: wortmannin. Values presented are means ± SEM. n = 8. *p < 0.05, **p < 0.01 vs. Sham, #p < 0.05 vs. MI/R, αp < 0.05 vs. MI/R+C.
Akt phosphorylation
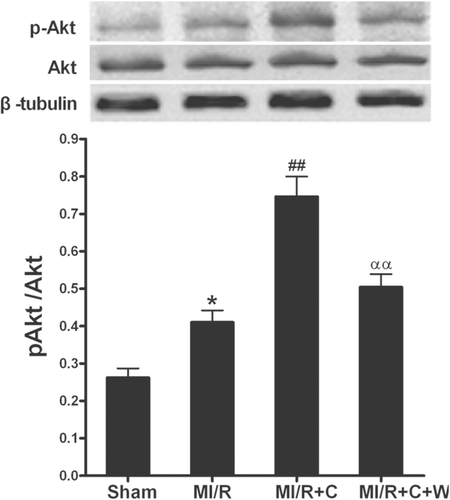
It has been proven that Akt activation exerts a beneficial effect on ischemic hearts (CitationXie et al., 2011). To further investigate the mechanisms underlying the catalpol-induced antioxidant/antinitrative effects, we measured the Akt expression and phosphorylation by Western blotting in myocardium in rats at the end of 3 h reperfusion (). There was no significant difference in Akt expression among different groups, while treatment with catalpol significantly increased the phosphorylation of Akt (p < 0.01). Pretreatment with wortmannin completely blocked Akt phosphorylation induced by catalpol. The results suggested that PI3K-Akt signaling pathway contributed to the beneficial effects of catalpol in MI/R.
Figure 7. Phosphorylation of Akt by in vivo catalpol treatment and its modification by wortmannin. Sham: sham-operated; MI/R: myocardial ischemia/reperfusion (30 min/3 h); C: catalpol, W: wortmannin. Values presented are means ± SEM. n = 6. *p < 0.05 vs. Sham, ##p < 0.01 vs. MI/R, ααp < 0.01 vs. MI/R+C.
Cardiac injury and nitrotyrosine content after inhibition of ONOO− formation
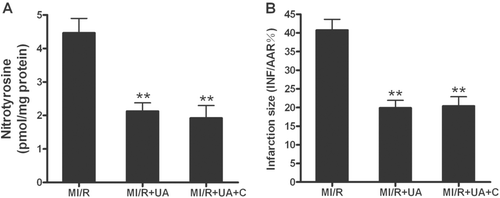
Our results suggest that catalpol may exert its cardioprotective effects by inhibition of the overproduction of ONOO−. To obtain more evidence to support this conclusion, an additional series of experiments was performed. Male adult rats were subjected to MI/R as described above and treated with either uric acid (5 mg/kg intravenous injection 10 min before ischemia), an ONOO− scavenger, or uric acid plus catalpol. As summarized in , treatment with uric acid significantly reduced MI/R-induced ONOO− production (p < 0.01). Co-treatment with catalpol had no additional effect (p > 0.05 vs. uric acid group). Uric acid treatment also markedly attenuated myocardial injury as determined by decreased myocardial infarct size (). Co-treatment with catalpol afforded no additional cardioprotection. These results demonstrated that scavenging ONOO− preferentially protected hearts against MI/R injury and that decreased production of ONOO− plays a critical role in the cardioprotection of catalpol.
Figure 8. Nitrotyrosine production (A) and infarction size (B) in rats subjected to MI/R with different treatments. MI/R: myocardial ischemia/reperfusion (30 min/3 h); UA: uric acid; C: catalpol. Values presented are means ± SEM. n = 6. **p < 0.01 vs. MI/R.
Discussion
Several important observations were made in our present experiments. First, we observed for the first time that catalpol has a novel protective effect against MI/R injury. Second, and most importantly, we demonstrated catalpol attenuates oxidative/nitrative stress and blocks the formation of toxic ONOO− in MI/R. Third, we provided direct evidence that inhibiting ·O2− overproduction and exerting an opposite effect on eNOS/iNOS activity through activating PI3K-Akt signaling pathway are the major mechanisms in catalpol-induced antioxidative/antinitrative effect.
Myocardial infarction remains the most prevalent cause of morbidity and mortality. Although rapid reperfusion of the occluded coronary arteries is ultimate treatment in salvaging ischemic myocardium and limiting the size of infarct, reperfusion may exacerbate cardiac injuries by production of ROS and ONOO−. Therefore, approaches need to seek to reduce MI/R-induced injury. Recently, catalpol has been identified as a vital protector of nervous system disease. Behavior studies exhibited that catalpol reversed brain damage and memory deficits in mice induced by lipopolysaccharide (LPS) and d-galactose and in gerbils by cerebral ischemia (Li et al., Citation2004b; CitationTian et al., 2006). Catalpol was also reported to protect against apoptosis in PC12 cells and dorsal hippocampus by suppression of Bax and upregulation of Bcl-2 (CitationLi et al., 2006). However, the effects of catalpol on MI/R-induced cardiac injury were still unknown. In the present study, we observed that MI/R rats showed severe myocardial functional impairment than normal state, which was consistent with the previous research. Catalpol treatment improved cardiac recovery as evidenced by improved cardiac functions and reduced infarct size, cell necrosis (decreased plasma CK and LDH activities) and apoptosis (reduced TUNEL-positive staining and attenuated capase-3 activity). These results suggested the beneficial effects of catalpol on I/R hearts.
There were numerous experimental and clinical investigations about ONOO− formation and its cytotoxic effects in myocardial ischemic/reperfused conditions (CitationWang & Zweier, 1996; CitationHayashi et al., 2001). ONOO− is a strong biological oxidant and nitrating species formed from the near-diffusion-limited reaction of the free radicals NO and ·O2−. The biological target of ONOO− involves proteins, lipids, and DNA (CitationPacher et al., 2005). ONOO− can also react with key components of mitochondria and thus causing peroxynitrite-mediated apoptotic and necrotic cell death (CitationLiang et al., 2010). Myocardial ONOO− formation and oxidative/nitrative stress have been shown to occur at 3 h after reperfusion (CitationJiao et al., 2009). The drugs that inhibit ONOO− formation or antagonize its toxicity could protect the heart from reperfusion injury (CitationJiao et al., 2009). These results provide direct evidence that ONOO− can be formed in I/R myocardium and contribute significantly to the post-ischemic myocardial injury. In the present study, catalpol treatment significantly decreased production of cardiac nitrotyrosine that has been widely used as a marker of nitrosative stress. It contributes to the decrease of myocardial injury and improvement of cardiac functional recovery following reperfusion. Interestingly, pretreatment with wortmannin markedly reversed catalpol-induced nitrotyrosine reduction. These data have provided the first evidence that the cardioprotection of catalpol after MI/R involves a novel mechanism of antioxidative/antinitrative stress by decreasing ONOO− overproduction through PI3K-dependent pathway.
The most interesting finding of the present study is that catalpol differentially regulates NO production from eNOS and iNOS in MI/R rats. eNOS phosphorylation and subsequent NO production have been well demonstrated to play antiapoptotic effect in ischemic/reperfused heart (CitationGao et al., 2002). Studies indicate that eNOS provides a constant, dynamic source of NO, whereas iNOS produces larger amounts of NO. Considerable evidence indicates that nitrative stress, which originated from the reaction between ROS and NO produced from iNOS in a diffusion-limited rate, plays a causative role in post-ischemic myocardial injury. Inhibiting iNOS activity or scavenging ONOO− may reduce nitrative stress and attenuate MI/R injury (CitationTao et al., 2007; CitationWang et al., 2007). In our present study, at the end of 3 h reperfusion (when iNOS is expressed in I/R myocardial tissue) I/R-induced iNOS expression was significantly inhibited after catalpol treatment; in contrast, catalpol treatment resulted in increased eNOS phosphorylation with a subsequent increase in NO production. Interestingly, wortmannin pretreatment inhibits catalpol-stimulated eNOS activation, NO production and iNOS reduction, and thus abolishes the catalpol-induced ONOO− reduction and cardioprotection. There are also report on catalpol’s effect in attenuating LPS-induced the expression of iNOS and thus exerting its protective effect on dopaminergic neurons (CitationTian et al., 2006; CitationLi et al., 2008). These data suggest that catalpol increases NO production from eNOS to exert the antiopototic effect of NO, and thus contributes to catalpol’s cardioprotective effect. On the other hand, under pathological conditions where iNOS expression is stimulated, catalpol inhibits iNOS expression and thus protects tissues from nitrative stress.
It has long been known that ROS can cause oxidative stress and inflict direct damage to I/R heart. Experimental evidence shows that ROS released during early phase of reperfusion strongly oxidize cardiomyocytes that have already been damaged by the ischemia (CitationMoens et al., 2005; CitationSaeed et al., 2005; CitationZweier & Talukder, 2006). Myocardial ROS generation during I/R has been shown to increase levels of inflammatory cytokines (such as TNF-α and IL-6). Inhibition of TNF-α potently blocks TNF-α-induced activation of NADPH oxidase (the most important source for ·O2− production) and resultant formation of nitrotyrosine in isolated coronary arterioles following I/R (CitationGao et al., 2008). All these suggest that inhibiting ·O2− generation may reduce ONOO− overproduction and resultant oxidative/nitrative stress. On the other hand, endogenous antioxidant pathways protect from increases in ROS, but are often compromised in MI/R states. An important antioxidant SOD, ·O2− scavenger, has been well demonstrated to reduce superoxide anion-dependent damages by transforming ·O2− into H2O2. In the present study, we have demonstrated that treatment with catalpol significantly reduced gp91phox (a critical component of NAPDH oxidase) expression and MDA (a marker of lipid peroxidation) in MI/R rats. In addition, catalpol treatment significantly improved total antioxidant capacity (SOD activity) in I/R heart. Pretreatment with wortmannin partly blocked catalpol’s effects. Consistent with our findings, CitationTian et al. (2006) demonstrated that catalpol significantly reduced the release of ROS and TNF-α to protected dopaminergic neurons against LPS-induced neurotoxicity. The increased ROS production and increased NO from iNOS favor the generation of ONOO−. Based on these observations, it is suggested that the reduction of ·O2− generation should be responsible for suppression of ONOO− afforded by catalpol in MI/R rats.
As to the mechanisms responsible for the antioxidative/antinitrative effects of catalpol in MI/R, here we observed that catalpol increased Akt activation. We have demonstrated that linolenic acid potently activated Akt in cultured cells and in vivo, which was proved to be a “prosurvival” molecule to inhibit cell apoptosis (Zhang et al., Citation2007b; CitationXie et al., 2011). Previous studies also showed that insulin exerts a cardioprotective effect against MI/R injury and apoptosis through activated PI3K-Akt-eNOS signaling, which can be reversed by wortmannin (CitationGao et al., 2002). We also observed that inhibition of PI3K with wortmannin pretreatment both blocked the Akt phosphorylation and antioxidative/antinitrative effects induced by catalpol, suggesting PI3K-Akt signaling contributes to the catalpol’s cardioprotection. Moreover, since PI3K-Akt is one of the main signaling pathways of insulin, catalpol could serve as an insulin sensitizer. Therefore, the future studies are expected to investigate the effect of catalpol on insulin resistance both locally and systemically.
NO itself is not toxic, but its potentially toxic effects depends on its conversion into higher nitrogen oxides (CitationKim et al., 1999; CitationPacher et al., 2007). The presence of ·O2− may divert NO to a toxic pathway by leading to the formation of ONOO−, which causes oxidative/nitrative stress and tissue injury (CitationBeckman et al., 1990). In our present study, we have demonstrated that catalpol treatment markedly reduced MI/R-induced ONOO− formation by inhibition of NADPH oxidase-derived ·O2− and iNOS-derived NO. In addition, scavenging ONOO− with UA preferentially also protected hearts against MI/R injury, whereas co-treatment with catalpol had no additional effect, which further supported that catalpol afforded cardioprotection via reducing ONOO−-caused oxidative/nitrative stress. Physiological or pharmacological concentrations of NO produced from eNOS or NO donors exert significant cardioprotective effects and antagonize the cytotoxic actions of ONOO−. Theoretically, a drug that possesses dual actions (i.e., stimulation of NO production and inhibition ·O2− of production) would provide the most protection against I/R injury. To our knowledge, catalpol is the one that provides this dual-protective effect and attenuates MI/R injury.
Conclusion
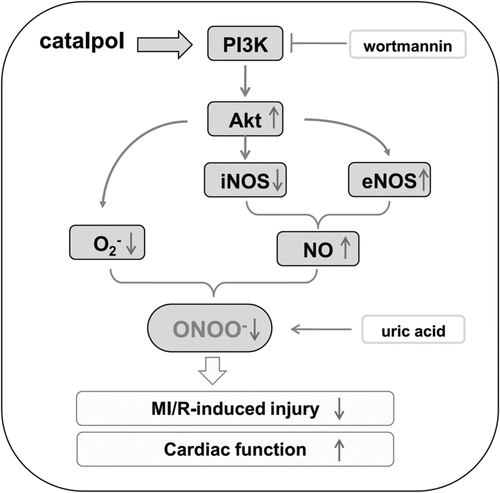
In summary, the data in the present study have demonstrated that catalpol attenuates oxidative/nitrative stress by blocking ONOO− formation. Increased physiological NO production (via eNOS phosphorylation) and ·O2− reduction through PI3K-Akt signaling pathway contribute to the antioxidative/antinitrative effects of catalpol ().
Figure 9. Proposed antioxidative/antinitrative signaling pathway by catalpol. eNOS: endothelial nitric oxide synthase; iNOS: inducible nitric oxide synthase; MI/R: myocardial ischemia/reperfusion; NO: nitric oxide; ·O2−: superoxide anion; ONOO−: peroxynitrite.
Declaration of interest
This study was supported by grants from the National Natural Science Foundation of China (Nos. 81270330, 81270329, 81100083, 81100165). The authors report no conflict of interest.
Related Research Data
References
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. (1990). Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA, 87, 1620–1624.
- Falk JA, Aune SE, Kutala VK, Kuppusamy P, Angelos MG. (2007). Inhibition of peroxynitrite precursors, NO and O2, at the onset of reperfusion improves myocardial recovery. Resuscitation, 74, 508–515.
- Gao F, Gao E, Yue TL, Ohlstein EH, Lopez BL, Christopher TA, Ma XL. (2002). Nitric oxide mediates the antiapoptotic effect of insulin in myocardial ischemia-reperfusion: The roles of PI3-kinase, Akt, and endothelial nitric oxide synthase phosphorylation. Circulation, 105, 1497–1502.
- Gao X, Zhang H, Belmadani S, Wu J, Xu X, Elford H, Potter BJ, Zhang C. (2008). Role of TNF-alpha-induced reactive oxygen species in endothelial dysfunction during reperfusion injury. Am J Physiol Heart Circ Physiol, 295, H2242–H2249.
- Hayashi Y, Sawa Y, Ohtake S, Fukuyama N, Nakazawa H, Matsuda H. (2001). Peroxynitrite formation from human myocardium after ischemia-reperfusion during open heart operation. Ann Thorac Surg, 72, 571–576.
- Ishida H, Ichimori K, Hirota Y, Fukahori M, Nakazawa H. (1996). Peroxynitrite-induced cardiac myocyte injury. Free Radic Biol Med, 20, 343–350.
- Jiao XY, Gao E, Yuan Y, Wang Y, Lau WB, Koch W, Ma XL, Tao L. (2009). INO-4885 [5,10,15,20-tetra[N-(benzyl-4′-carboxylate)-2-pyridinium]-21H,23H-porphine iron(III) chloride], a peroxynitrite decomposition catalyst, protects the heart against reperfusion injury in mice. J Pharmacol Exp Ther, 328, 777–784.
- Kim YM, Bombeck CA, Billiar TR. (1999). Nitric oxide as a bifunctional regulator of apoptosis. Circ Res, 84, 253–256.
- Levrand S, Vannay-Bouchiche C, Pesse B, Pacher P, Feihl F, Waeber B, Liaudet L. (2006). Peroxynitrite is a major trigger of cardiomyocyte apoptosis in vitro and in vivo. Free Radic Biol Med, 41, 886–895.
- Li DQ, Bao YM, Li Y, Wang CF, Liu Y, An LJ. (2006). Catalpol modulates the expressions of Bcl-2 and Bax and attenuates apoptosis in gerbils after ischemic injury. Brain Res, 1115, 179–185.
- Li DQ, Bao YM, Zhao JJ, Liu CP, Liu Y, An LJ. (2004a). Neuroprotective properties of catalpol in transient global cerebral ischemia in gerbils: Dose-response, therapeutic time-window and long-term efficacy. Brain Res, 1029, 179–185.
- Li DQ, Duan YL, Bao YM, Liu CP, Liu Y, An LJ. (2004b). Neuroprotection of catalpol in transient global ischemia in gerbils. Neurosci Res, 50, 169–177.
- Li R, Wang WQ, Zhang H, Yang X, Fan Q, Christopher TA, Lopez BL, Tao L, Goldstein BJ, Gao F, Ma XL. (2007). Adiponectin improves endothelial function in hyperlipidemic rats by reducing oxidative/nitrative stress and differential regulation of eNOS/iNOS activity. Am J Physiol Endocrinol Metab, 293, E1703–E1708.
- Li Y, Bao Y, Jiang B, Wang Z, Liu Y, Zhang C, An L. (2008). Catalpol protects primary cultured astrocytes from in vitro ischemia-induced damage. Int J Dev Neurosci, 26, 309–317.
- Liang JH, Li YN, Qi JS, Jia XX. (2010). Peroxynitrite-induced protein nitration is responsible for renal mitochondrial damage in diabetic rat. J Endocrinol Invest, 33, 140–146.
- Lopez BL, Liu GL, Christopher TA, Ma XL. (1997). Peroxynitrite, the product of nitric oxide and superoxide, causes myocardial injury in the isolated perfused rat heart. Coron Artery Dis, 8, 149–153.
- Ma XL, Lopez BL, Liu GL, Christopher TA, Ischiropoulos H. (1997). Peroxynitrite aggravates myocardial reperfusion injury in the isolated perfused rat heart. Cardiovasc Res, 36, 195–204.
- Moens AL, Claeys MJ, Timmermans JP, Vrints CJ. (2005). Myocardial ischemia/reperfusion-injury, a clinical view on a complex pathophysiological process. Int J Cardiol, 100, 179–190.
- Pacher P, Beckman JS, Liaudet L. (2007). Nitric oxide and peroxynitrite in health and disease. Physiol Rev, 87, 315–424.
- Pacher P, Schulz R, Liaudet L, Szabó C. (2005). Nitrosative stress and pharmacological modulation of heart failure. Trends Pharmacol Sci, 26, 302–310.
- Saeed SA, Waqar MA, Zubairi AJ, Bhurgri H, Khan A, Gowani SA, Waqar SN, Choudhary MI, Jalil S, Zaidi AH, Ara I. (2005). Myocardial ischaemia and reperfusion injury: reactive oxygen species and the role of neutrophil. J Coll Physicians Surg Pak, 15, 507–514.
- Su H, Sun X, Ma H, Zhang HF, Yu QJ, Huang C, Wang XM, Luan RH, Jia GL, Wang HC, Gao F. (2007). Acute hyperglycemia exacerbates myocardial ischemia/reperfusion injury and blunts cardioprotective effect of GIK. Am J Physiol Endocrinol Metab, 293, E629–E635.
- Tao L, Gao E, Jiao X, Yuan Y, Li S, Christopher TA, Lopez BL, Koch W, Chan L, Goldstein BJ, Ma XL. (2007). Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation, 115, 1408–1416.
- Tian YY, An LJ, Jiang L, Duan YL, Chen J, Jiang B. (2006). Catalpol protects dopaminergic neurons from LPS-induced neurotoxicity in mesencephalic neuron-glia cultures. Life Sci, 80, 193–199.
- Virág L, Szabó E, Gergely P, Szabó C. (2003). Peroxynitrite-induced cytotoxicity: Mechanism and opportunities for intervention. Toxicol Lett, 140-141, 113–124.
- Wang P, Zweier JL. (1996). Measurement of nitric oxide and peroxynitrite generation in the postischemic heart. Evidence for peroxynitrite-mediated reperfusion injury. J Biol Chem, 271, 29223–29230.
- Wang XL, Liu HR, Tao L, Liang F, Yan L, Zhao RR, Lopez BL, Christopher TA, Ma XL. (2007). Role of iNOS-derived reactive nitrogen species and resultant nitrative stress in leukocytes-induced cardiomyocyte apoptosis after myocardial ischemia/reperfusion. Apoptosis, 12, 1209–1217.
- Xie N, Zhang W, Li J, Liang H, Zhou H, Duan W, Xu X, Yu S, Zhang H, Yi D. (2011). a-Linolenic acid intake attenuates myocardial ischemia/reperfusion injury through anti-inflammatory and anti-oxidative stress effects in diabetic but not normal rats. Arch Med Res, 42, 171–181.
- Zhang HF, Fan Q, Qian XX, Lopez BL, Christopher TA, Ma XL, Gao F. (2004). Role of insulin in the anti-apoptotic effect of glucose-insulin-potassium in rabbits with acute myocardial ischemia and reperfusion. Apoptosis, 9, 777–783.
- Zhang KR, Liu HT, Zhang HF, Zhang QJ, Li QX, Yu QJ, Guo WY, Wang HC, Gao F. (2007a). Long-term aerobic exercise protects the heart against ischemia/reperfusion injury via PI3 kinase-dependent and Akt-mediated mechanism. Apoptosis, 12, 1579–1588.
- Zhang W, Wang R, Han SF, Bu L, Wang SW, Ma H, Jia GL. (2007b). Alpha-linolenic acid attenuates high glucose-induced apoptosis in cultured human umbilical vein endothelial cells via PI3K/Akt/eNOS pathway. Nutrition, 23, 762–770.
- Zweier JL, Talukder MA. (2006). The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res, 70, 181–190.