Abstract
Context: In the course of searching potential antitumor agents from a library of chalcones synthesized under microwave irradiations, the brine shrimp lethality (BSL) assay and a 3D structure–activity relationship (3DQSAR) studies were followed by the antitumor evaluation of most potent analogues.
Objective: The objective of the current study was to effectively use the BSL assay for the identification of potential cytotoxic analogues from a set of compounds.
Methods: We applied the comparative molecular field analysis (CoMFA) and devised 3DQSAR on 33 synthesized chalcones leading to prediction of five related compounds with improved activity. The scope of BSL assay for the prediction of antitumor potency was tested through the in vitro antitumor studies against six human tumor cell-lines, docking studies and the tubulin-polymerization assay.
Results: The newly designed compounds 34–38 displayed very promising cytotoxic potency. From our results, the BSL toxicity, antitumor efficacy and docking outcomes could be easily co-related.
Conclusion: The study draws a very good relationship between a simple, inexpensive, and bench-top BSL assay and the antitumor potential of the cytotoxic compounds. Devising the CoMFA analysis helped in designing chalcones with improved cytotoxic potential as displayed through their BSL and cytotoxic activity against human tumor cell lines. The studies are noteworthy as such comprehensive studies were never performed before on the BSL assay. The present studies widen the scope of the BSL model that may prove quite helpful as a preliminary screen in the antitumor drug designing and synthesis expeditions.
Introduction
Chalcones are bioactive and aromatic flavonoids found naturally in plants. They are mostly non-toxic, show compliance with the Rule of Five (Ro5), and therefore are expected to be bioavailable. Chalcones are gaining importance for their medicinal value and are extensively investigated for their pharmacological importance. They have shown an impressive array of biological activities, including anti-inflammatory (Furusawa et al., Citation2009; Won et al., Citation2005; Yang et al., Citation2007), antitumor (Ahsanullah et al., Citation2007; Aryapour et al., Citation2012; Ducki, Citation2009; Dyrager et al., Citation2011; Kong et al., Citation2010; Na et al., Citation2009; Zhou et al., Citation2009), antioxidant (Gacche et al., Citation2008; Ghosh et al., Citation2009), antiviral (Deng et al., Citation2007; Kiat et al., Citation2006), antibacterial (Ansari et al., Citation2005, Citation2009; Sivakumar et al., Citation2007, Citation2009), antileshmanial (Boeck et al., Citation2006; Kayser & Kiderlen, Citation2001; Liu et al., Citation2003), antimalarial (Dominguez et al., Citation2005a,Citationb; Geyer et al., Citation2009), antihyperglycemic (Damazio et al., Citation2010) and antituberculotic (Lin et al., Citation2002). They are also patented as antiangiogenic agents (Bowen et al., Citation2005), matrix metalloproteinases (MMP’s) inhibitors (Kim et al., Citation2003) and as anticancer agents (Cai et al., Citation2007).
In this study, we have attempted to use comparative molecular field analysis (CoMFA), a designing paradigm, for the 3-dimensional structure–activity relationship (3D-QSAR) and subsequent designing of potent cytotoxic chalcones. The preliminary cytotoxic activities were estimated by the brine shrimp lethality (BSL) assay. Subsequently, five new compounds with improved cytotoxic activity were designed based on the initial data of 33 chalcones. Their pLC50 values were predicted from the derived QSAR equation. The cytotoxicity was further evaluated through docking studies with tubulin protein and free energies were calculated. The compounds were then analyzed in vitro for their cytotoxic activity against six human cancer cell lines. The mode of action was also verified by the tubulin polymerization assay. We report that the BSL assay based the in silico designing and synthesis of compounds may help in finding potent antitumor drugs.
Materials and methods
Chemicals and instruments
The commercially available chemicals acetophenonenes, benzaldehydes, sodium hydroxide, RPMI 1640 medium, DMEM medium, 1,4-piperazinediethane sulfonic acid, EGTA, MgCl2, GTP, glycerol, n-hexane, ethyl acetate, ethanol and dichloromethane were obtained from Sigma-Aldrich (St. Louis, MO) and were used without further purification. Taxol and colchicine were purchased from Tocris Bioscience (Ellisville, MO). Lyophilized bovine tubulin was obtained from Cytoskeleton Inc (St. Denver, CO). The 96-well flat-bottom plates were obtained from Corning Costar (Lowell, MA). Solution-phase microwave synthesis was performed in 200 mL round-bottom flasks using a household microwave oven, 900 W, Model No. MS-304A, LG (Seoul, Korea). Water was used as a heat sink. Thin-layer chromatography (TLC) was performed on Merck (Darmstadt, Germany) silica gel HF254 plates (0.25 mm). Melting points were determined in open capillaries on a Mel-Temp apparatus and are uncorrected. IR spectra were recorded on a Schimadzu FTIR-270 spectrophotometer using KBr pellets. 1H NMR spectra were recorded on NMR Bruker (Billerica, MA) apparatus at 300 and 400 MHz in CDCl3. Tetramethylsilane (TMS) was used as internal reference. Chemical shifts are given in parts per million (ppm). HR-MS spectra were recorded on Varian MAT 311A, Varian MAT GmbH (Bremen, Germany). Elemental analysis was performed on a CHN analyzer 932 provided by Leco (St. Joseph, MI). Some of the compounds synthesized were purified through flash chromatography using silica gel; Kieselgel G-60 (Merck). Bioassays were performed on the PowerWave HT microplate reader, Bio-Tek Instruments (Highland Park, VT).
General procedure for the synthesis of chalcones (1–38)
To an ethanol solution (15 mL) of acetophenone (5 mmol) and benzaldehyde (5 mmol), 4.0 M aq. NaOH soln. (30 mL) was added, and the mixture was irradiated in a microwave oven till the reaction was found to be complete (Ansari et al., Citation2005). The mixture was kept at 0–4 °C overnight, neutralized with ice-cold dil. aq. HCl. The resulting precipitate was filtered off and purified either by recrystallization from EtOH or by flash chromatography (SiO2; hexane/AcOEt). The purity of the compounds was checked by multiple TLC (SiO2; hexane:AcOEt 3:1, 2:1 and 1:1).
(E)-1,3-(Diphenyl)-2-propen-1-one (1)
Compound 1 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and benzaldehyde (0.507 mL, 5 mmol). Yield 94%. m.p. 56–58 °C. (KBr): ν = 3076, 1656, 1600, 1598. 1H NMR (300 MHz, DMSO-d6): δ = 7.46 (bt, J = 3 Hz, 3H, H–C(3,4,5)), 7.57 (t, J = 7.33 Hz, 2H, H–C(3′,5′)), 7.67 (t, J = 7.32 Hz, 1H, H–C(4′)), 7.74 (d, 1H, J = 15.9 Hz, H–Cα(vinyl)), 7.95 (m, 3H, H–Cβ(vinyl), H–C(2,6)), 8.15 (d, 1H, J = 7.33 Hz, H–C(2′,6′)).
(E)-1-(Phenyl)-3-(2-hydroxyphenyl)-2-propen-1-one (2)
Compound 2 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and 2-hydroxybenzaldehyde (0.388 mL, 5 mmol). Yield 67%. m.p. 48–50 °C. IR (KBr): ν = 3600–3100, 1663, 1605, 1575. 1H NMR (300 MHz, DMSO-d6): δ = 7.46 (t, J = 3.2 Hz, 2H, H–C(3,6)), 7.57 (t, J = 7.5 Hz, 2H, H–C(4,5)), 7.67 (t, J = 7.6 Hz, 1H, H–C(4′)), 7.72 (d, J = 15.2 Hz, 1H, H–Cα(vinyl)), 7.87–7.96 (m, 2H, H–Cβ(vinyl), H–C(3′,5′)), 8.15 (d, J = 8.1 Hz, 1H, H–C(2′,6′)), 10.38 (s, 1H, H–O).
(E)-1-(Phenyl)-3-(2-methoxyphenyl)-2-propen-1-one (3)
Compound 3 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and 2-methoxybenzaldehyde (0.604 mL, 5 mmol). Yield 65%. m.p. 75–77 °C. IR (KBr): ν = 3055, 2940, 1651, 1600, 1595. 1H NMR (300 MHz, DMSO-d6): δ = 3.80 (s, 3H, H–C(OCH3)), 7.01 (d, J = 8.5 Hz, 2H, H–C(3,6)), 7.56 (bt, J = 6.11 Hz, 2H, H–C(4,5)), 7.62–7.73 (m, 2H, H–Cα(vinyl), H–C(4′)), 7.80 (d, J = 15.8 Hz, 1H, H–Cβ(vinyl)), 7.85 (d, J = 8.5 Hz, 2H, H–C(3′,5′)). 8.13 (d, J = 6.1 Hz, 2H, H–C(2′,6′)).
(E)-1-(Phenyl)-3-(3-methoxyphenyl)-2-propen-1-one (4)
Compound 4 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and 3-methoxybenzaldehyde (0.609 mL, 5 mmol). Yield 65%. m.p. 56–58 °C. IR (KBr): ν = 3034, 2973, 1637, 1587. 1H NMR (400 MHz, CD3OD): δ = 3.83 (s, 3H, H–C(OCH3)), 6.98 (ddd, J = 8.1 Hz, 1.6 Hz, 1.5 Hz, 1H, H–C(4)), 7.24 (dd, J = 1.9 Hz, 1.4 Hz, 1H, H–C(2)), 7.29–7.34 (m, 2H, H–C(5,6)), 7.52 (ddd, J = 7.8 Hz, 7.3 Hz, 1.4 Hz, 2H, H–C(3′,5′)), 7.61–7.63 (m, 1H, H–C(4′)), 7.66 (d, J = 15.6 Hz, 1H, H–Cα(vinyl)), 7.74 (d, J = 15.8 Hz, 1H, H–Cβ(vinyl)), 8.05 (ddd, J = 7.0 Hz, 1.9 Hz, 1.4 Hz, 2H, H–C(2′,6′)).
(E)-1-(Phenyl)-3-(4-methoxyphenyl)-2-propen-1-one (5)
Compound 5 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and 4-methoxybenzaldehyde (0.432 mL, 5 mmol). Yield 82%. m.p. 74–76 °C. IR (KBr): ν = 3034, 2895, 1651, 1596. 1H NMR (400 MHz, CD3OD): δ = 3.84 (s, 3H, H–C(OCH3)), 6.97 (dd, J = 7.0 Hz, 1.9, 2H, H–C(3,5)), 7.21 (dd, J = 7.0 Hz, 1.9 Hz, 2H, H–C(2,6)), 7.50–7.54 (m, 2H, H–C(3′,5′)), 7.59 (d, J = 15.5 Hz, 1H, H–Cα(vinyl)), 7.65–7.68 (m, 1H, H–C(4′)), 7.75 (d, J = 15.5 Hz, 1H, H–Cβ(vinyl)), 8.04 (ddd, J = 7.3 Hz, 1.9 Hz, 1.4 Hz, 2H, H–C(2′,6′)).
(E)-1-(Phenyl)-3-(2-nitrophenyl)-2-propen-1-one (6)
Compound 6 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and 2-nitrobenzaldehyde (0.775 g, 5 mmol). Yield 65%. m.p. 86–88 °C. IR (KBr): ν = 3072, 1674, 1517, 1472, 1441. Anal. Calcd: C, 71.14%, H, 4.38%, N, 5.53%. Found: C, 71.26%, H, 4.43%, N, 5.51%.
(E)-1-(Phenyl)-3-(3-nitrophenyl)-2-propen-1-one (7)
Compound 7 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and 3-nitrobenzaldehyde (0.775 g, 5 mmol). Yield 83%. m.p. 119–121 °C. IR (KBr): ν = 3074, 1690, 1592, 1489, 1441. Anal. Calcd: C, 71.14%, H, 4.38%, N, 5.53%. Found: C, 71.22%, H, 4.40%, N, 5.52%.
(E)-1-(Phenyl)-3-(4-nitrophenyl)-2-propen-1-one (8)
Compound 8 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and 4-nitrobenzaldehyde (0.775 g, 5 mmol). Yield 60%. m.p. 145–146 °C. IR (KBr): ν = 3065, 1670, 1508, 1467, 1331. Anal. Calcd: C, 71.14%, H, 4.38%, N, 5.53%. Found: C, 71.22%, H, 4.36%, N, 5.51%.
(E)-1-(Phenyl)-3-(2-chlorophenyl)-2-propen-1-one (9)
Compound 9 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and 2-chlorobenzaldehyde (0.417 mL, 5 mmol). Yield 78%. m.p. 45–46 °C. IR (KBr): ν = 3055, 1652, 1600, 1770. 1H NMR (400 MHz, CD3OD): δ = 7.08 (dt, J = 7.5 Hz, 1.5 Hz, 1H, H–C(4)), 7.10–7.12 (m, 1H, H–C(5)), 7.22 (dd, J = 7.4 Hz, 1.4 Hz, 1H, H–C(3)), 7.24 (dd, J = 7.5 Hz, 1.7 Hz, 1H, H–C(6)), 7.39 (d, J = 14.8 Hz, 1H, H–Cα(vinyl)), 7.36–7.53 (m, 5H, H–C(arom.)), 8.17 (d, J = 14.9 Hz, 1H, H–Cβ(vinyl)). Anal. Calcd: C, 74.23%, H, 4.57%. Found: C, 73.93%, H, 4.58%.
(E)-1-(Phenyl)-3-(3-chlorophenyl)-2-propen-1-one (10)
Compound 10 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and 3-chlorobenzaldehyde (0.506 mL, 5 mmol). Yield 81%. m.p. 68–70 °C. IR (KBr): ν = 3061, 1645, 1583, 1554, 689. Anal. Calcd: C, 74.23%, H, 4.57%. Found: C, 74.18%, H, 4.57%.
(E)-1-(Phenyl)-3-(4-chlorophenyl)-2-propen-1-one (11)
Compound 11 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and 4-chlorobenzaldehyde (0.652 g, 5 mmol). Yield 82%. m.p. 114–116 °C. IR (KBr): ν = 3086, 1655, 1600, 761. Anal. Calcd: C, 74.23%, H, 4.57%. Found: C, 74.30%, H, 4.61%.
(E)-1-(Phenyl)-3-(2-bromophenyl)-2-propen-1-one (12)
Compound 12 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and 2-bromobenzaldehyde (0.584 mL, 5 mmol). Yield 83%. m.p. 98–100 °C. IR (KBr): ν = 3059, 1683, 1593, 1567, 751. Anal. Calcd: C, 62.74%, H, 3.86%. Found: C, 62.69%, H, 3.86%.
(E)-1-(Phenyl)-3-(3-bromophenyl)-2-propen-1-one (13)
Compound 13 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and 3-bromobenzaldehyde (0.580 mL, 5 mmol). Yield 85%. m.p. 82–84 °C. IR (KBr): ν = 3061, 1658, 1601, 1556, 769. Anal. Calcd: C, 62.74%, H, 3.86%. Found: C, 62.55%, H, 3.89%.
(E)-1-(Phenyl)-3-(4-methylphenyl)-2-propen-1-one (14)
Compound 14 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and 4-methylbenzaldehyde (0.587 mL, 5 mmol). Yield 79%. m.p. 92–94 °C. IR (KBr): ν = 3079, 2710, 1650, 1600, 1595. 1H NMR (400 MHz, CD3OD): δ = 2.35 (s, 3H, H–C(CH3)), 7.01 (dd, J = 7.9 Hz, 1.6 Hz, 2H, H–C(3,5)), 7.18 (dd, J = 7.7 Hz, 1.5 Hz, 2H, H–C(2,6)), 7.47 (dt, J = 8.0 Hz, 1.4 Hz, 2H, H–C(3′,5′)), 7.60 (m, 1H, H–C(4′)), 7.67 (d, J = 15.7 Hz, 1H, H–Cα(vinyl)), 7.80 (dd, J = 8.1 Hz, 1.4 Hz, 2H, H–C(2′,6′)), 7.87 (d, J = 15.7 Hz, 1H, H–Cβ(vinyl)). Anal. Calcd: C, 86.45%, H, 6.35%. Found: C, 86.66%, H, 6.61%.
(E)-1-(Phenyl)-3-(3,4-dimethoxyphenyl)-2-propen-1-one (15)
Compound 15 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and 3,4-dimethoxybenzaldehyde (0.830 g, 5 mmol). Yield 73%. m.p. 70–71 °C. IR (KBr): ν = 3041, 2914, 1651, 1600, 1595. 1H NMR (400 MHz, CD3OD): δ = 3.84 (s, 6H, H–C(OCH3)), 6.98 (d, J = 1.5 Hz, 1H, H–C(2)), 7.29 (dd, J = 9.3 Hz, 1.7 Hz, 1H, H–C(6)), 7.36 (d, J = 9.1 Hz, 1H, H–C(5)), 7.52 (ddd, J = 7.7 Hz, 7.3 Hz, 1.5 Hz, 2H, H–C(3′,5′)), 7.61 (d, J = 15.6 Hz, 1H, H–Cα(vinyl)), 7.73 (d, J = 15.5 Hz, 1H, H–Cβ(vinyl)), 7.81 (ddd, J = 7.7 Hz, 1.5 Hz, 1.3 Hz, 2H, H–C(2′,6′)), 7.57–7.59 (m, 1H, H–C(4′)).
(E)-1-(4′-Aminophenyl)-3-(phenyl)-2-propen-1-one (16)
Compound 16 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5 mmol) and benzaldehyde (0.507 mL, 5 mmol). Yield 68%. m.p. 84–86 °C. IR (KBr): ν = 3041, 2847, 2555, 1659, 1597, 1576. 1H NMR (400 MHz, CDCl3): δ = 3.4 (bs, 2H, H–N(NH2)), 7.02 (d, J = 16.0 Hz, 1H, H–Cα(vinyl)), 7.06 (d, J = 16.0 Hz, 1H, H–Cβ(vinyl)), 7.33–7.34 (m, 4H, H–C(2′,6′,3′,5′)), 7.57–7.61 (m, 5H, H–C(arom.). HRMS: Calcd [(M+) = C15H13NO] 223.0998; found 223.0993 m/z (88%).
(E)-1-(4′-Aminophenyl)-3-(2-hydroxyphenyl)-2-propen-1-one (17)
Compound 17 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5 mmol) and 2-hydroxybenzaldehyde (0.388 mL, 5 mmol). Yield 67%. m.p. 104–108 °C. IR (KBr): ν = 3327, 3259, 3070, 3062, 2380, 1676, 1584, 1574. 1H NMR (400 MHz, CDCl3): δ = 5.98 (bs, 2H, H–N(NH2)), 6.92 (t, J = 8.0 Hz, 1H, H–C(4)), 6.98 (m, 2H, H–Cα(vinyl), H–C(5)), 7.04 (d, J = 8.1 Hz, 1H, H–C(6)), 7.31 (d, J = 8.3 Hz, 2H, H–C(3′,5′)), 7.41 (d, J = 8.9 Hz, 1H, H–C(3)), 7.75 (d, J = 14.8 Hz, 1H, H–Cβ(vinyl)), 8.01 (d, J = 8.3 Hz, 2H, H–C(2′,6′)), 9.88 (s, 1H, H–O(OH)). HRMS: Calcd [(M+) = C15H13NO2] 239.0947; found 239.0939 m/z (83.8%).
(E)-1-(4′-Aminophenyl)-3-(3-hydroxyphenyl)-2-propen-1-one (18)
Compound 18 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5 mmol) and 3-hydroxybenzaldehyde (0.610 g, 5 mmol). Yield 65%. m.p. 197 °C (decomp.). IR (KBr): ν = 3380–3120, 3327, 3259, 3062, 1676, 1584, 1573. 1H NMR (400 MHz, (CDCl3): δ = 4.1 (bs, 2H, H–N(NH2)), 6.68 (d, J = 8.6 Hz, 1H, H–C(6)), 6.80 (d, J = 8.2 Hz, 2H, H–C(3′,5′)), 6.87 (t, J = 8.7 Hz, 1H, H–C(5)), 7.09 (s, 1H, H–C(2)), 7.43 (d, J = 15.4 Hz, 1H, H–Cα(vinyl)), 7.51 (d, J = 8.8 Hz, 1H, H–C(4)), 7.73 (d, J = 15.4 Hz, 1H, H–Cβ(vinyl)), 8.03 (d, J = 8.2 Hz, 2H, H–C(2′,6′)), 9.76 (s, 1H, H–O(OH)). HRMS: Calcd [(M+ ) = C15H13NO2] 239.0936; found 239.0947 m/z (80.9%).
(E)-1-(4′-Aminophenyl)-3-(2-methoxyphenyl)- 2-propen-1-one (19)
Compound 19 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5 mmol) and 2-methoxybenzaldehyde (0.604 mL, 5 mmol). Yield 77%. m.p. 105–107 °C. IR (KBr): ν = 3440, 3349, 3241, 3237, 1639, 1605, 1549. 1H NMR (400 MHz, CDCl3): δ = 3.46 (s, 3H, H–C(OCH3)), 3.80 (bs, 2H, H–N(NH2)), 6.90 (bd, J = 7.6 Hz, 2H, H–C(3,6)), 7.39 (d, J = 15.6 Hz, 1H, H–Cα(vinyl)), 7.60 (bt, J = 7.7 Hz, 2H, H–C(4,5)), 7.73 (d, J = 15.6 Hz, 1H, H–Cβ(vinyl)), 7.90 (d, J = 8.2 Hz, 2H, H–C(2′,6′)), 8.69 (d, J = 8.2 Hz, 2H, H–C(3′,5′)). HRMS: calcd [(M+) = C16H15NO2] 253.1104; found 253.1091 m/z (80.5%).
(E)-1-(4′-Aminophenyl)-3-(3-methoxyphenyl)- 2-propen-1-one (20)
Compound 20 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5 mmol) and 3-methoxybenzaldehyde (0.608 mL, 5 mmol). Yield 73%. m.p. 134 °C. IR (KBr): ν = 3466, 3329, 3214, 1630, 1600, 1597. 1H NMR (400 MHz, CDCl3): δ = 3.63 (s, 3H, H–C(OCH3)), 4.13 (bs, 2H, H–N(NH2)), 6.68 (d, J = 8.0 Hz, 2H, H–C(3′,5′)), 6.92 (d, J = 7.4 Hz, 1H, H–C(6)), 7.22 (d, J = 7.5 Hz, 1H, H–C(4)), 7.23 (s, 1H, H–C(2)), 7.30 (t, J = 7.5 Hz, 1H, H–C(5)), 7.50 (d, J = 15.4 Hz, 1H, H–Cα(vinyl)), 7.76 (d, J = 15.4 Hz, 1H, H–Cβ(vinyl)), 7.90 (d, J = 8.0 Hz, 2H, H–C(2′,6′)). HRMS: Calcd [(M+) = C16H15NO2] 253.1104; found 253.1089 m/z (89.4%).
(E)-1-(4′-Aminophenyl)-3-(4-methoxyphenyl)- 2-propen-1-one (21)
Compound 21 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5 mmol) and 4-methoxybenzaldehyde (0.252 mL, 5 mmol). Yield 75%. m.p. 128–130 °C. IR (KBr): ν = 3449, 3354, 3245, 1646, 1604, 1551. 1H NMR (400 MHz, CDCl3): δ = 3.83 (s, 3H, H–C(OCH3)), 4.12 (bs, 2H, H–N(NH2)), 6.68 (d, J = 8.2 Hz, 2H, H–C(3′,5′)), 6.92 (d, J = 7.6 Hz, 2H, H–C(2,6)), 7.23 (d, J = 7.6 Hz, 2H, H–C(3,5)), 7.49 (d, J = 16 Hz, 1H, H–Cα(vinyl)), 7.72 (d, J = 16 Hz, 1H, H–Cβ(vinyl)), 7.90 (d, J = 8.2 Hz, 2H, H–C(2′,6′)). HRMS: Calcd [(M+) = C16H15NO2] 253.1104; found 253.1093 m/z (77.8%).
(E)-1-(4′-Aminophenyl)-3-(2-nitrophenyl)- 2-propen-1-one (22)
Compound 22 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5 mmol) and 2-nitrobenzaldehyde (0.776 g, 5 mmol). Yield 65%. m.p. 164–166 °C. IR (KBr): ν = 3474, 3382, 3227, 1665, 1515, 1482, 1412. 1H NMR (400 MHz, CDCl3): δ = 3.9 (bs, 2H, H–N(NH2)), 6.79 (d, J = 8.3 Hz, 2H, H–C(3′,5′)), 7.45 (t, J = 7.8 Hz, 1H, H–C(4)), 7.67 (t, J = 7.6 Hz, 1H, H–C(5)), 7.79 (d, J = 7.5 Hz, 1H, H–C(6)), 7.83 (d, J = 14.0 Hz, 1H, H–Cα(vinyl)), 8.02 (d, J = 8.3 Hz, 2H, H–C(2′,6′)), 8.13 (d, J = 8.0 Hz, 1H, H–C(3)), 8.22 (d, J = 14.0 Hz, 1H, H–Cβ(vinyl)). HRMS: Calcd [(M+) = C15H12N2O3] 268.0849; found 268.0847 m/z (11.8%).
(E)-1-(4′-Aminophenyl)-3-(3-nitrophenyl)- 2-propen-1-one (23)
Compound 23 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5 mmol) and 3-nitrobenzaldehyde (0.776 g, 5 mmol). Yield 83%. m.p. 206–207 °C. IR (KBr): ν = 3469, 3380, 3227, 1690, 1595, 1514, 1441. 1H NMR (400 MHz, CDCl3): δ = 4.20 (bs, 2H, H–N(NH2)), 6.79 (d, J = 8.0 Hz, 2H, H–C(3′,5′)), 6.90 (d, J = 6.1 Hz, 1H, H–C(6)), 7.60 (bt, J = 7.3 Hz, 1H, H–C(5)), 7.62 (d, J = 16.4 Hz, 1H, H–Cα(vinyl)), 7.78 (d, J = 16.4 Hz, 1H, H–Cβ(vinyl)), 8.03 (d, J = 8.0 Hz, 2H, H–C(2′,6′)), 8.24 (d, J = 8.4 Hz, 1H, H–C(4)), 8.49 (s, 1H, H–C(2)). HRMS: Calcd [(M+) = C15H12N2O3] 268.0849; found 268.0846 m/z (18.8%).
(E)-1-(4′-Aminophenyl)-3-(4-nitrophenyl)- 2-propen-1-one (24)
Compound 24 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5 mmol) and 4-nitrobenzaldehyde (0.776 g, 5 mmol). Yield 87%. m.p. 204–205 °C. IR (KBr): ν = 3481, 3385, 3228, 1670, 1508, 1478, 1407. 1H NMR (400 MHz, CDCl3): δ = 4.28 (bs, 2H, H–N(NH2)), 7.62 (d, J = 15.4 Hz, 1H, H–Cα(vinyl)), 7.76 (d, J = 15.4 Hz, 1H, H–Cβ(vinyl)), 7.92 (d, J = 8.4 Hz, 2H, H–C(3′,5′)), 8.03 (d, J = 8.4 Hz, 2H, H–C(2′,6′)), 8.24 (d, J = 8.8 Hz, 2H, H–C(2,6)), 8.28 (d, J = 8.8 Hz, 2H, H–C(3,5)). HRMS: Calcd [(M+) = C15H12N2O3] 268.0849; found 268.0844 m/z (12.5%).
(E)-1-(4′-Aminophenyl)-3-(2-chlorophenyl)- 2-propen-1-one (25)
Compound 25 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5 mmol) and 2-chlorobenzaldehyde (0.416 mL, 5 mmol). Yield 78%. m.p. 129 °C. IR (KBr): ν = 3452, 3351, 3243, 1654, 1582, 1558, 850. 1H NMR (400 MHz, CDCl3): δ = 4.13 (bs, 2H, H–N(NH2)), 6.68 (d, J = 8.3 Hz, 2H, H–C(3′,5′)), 7.28 (bt, J = 8.6 Hz, 2H, H–C(4,5)), 7.41 (d, J = 8.7 Hz, 1H, H–C(6)), 7.47 (d, J = 15.7 Hz, 1H, H–Cα(vinyl)), 7.71 (d, J = 8.5 Hz, 1H, H–C(3)), 7.90 (d, J = 8.3 Hz, 2H, H–C(2′,6′)), 8.11 (d, J = 15.7 Hz, 1H, H–Cβ(vinyl)). HRMS: Calcd [(M+) = C15H12NOCl35] 257.0608; found 257.0595 m/z (29.9%), calcd [(M+) = C15H12NOCl37] 259.0567; found 259.0556 m/z (9.9%).
(E)-1-(4′-Aminophenyl)-3-(3-chlorophenyl)- 2-propen-1-one (26)
Compound 26 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5 mmol) and 3-chlorobenzaldehyde (0.526 mL, 5 mmol). Yield 81%. m.p. 146 °C (decomp.). IR (KBr): ν = 3448, 3354, 3244, 1645, 1583, 1554, 869. 1H NMR (400 MHz, CDCl3): δ = 4.15 (bs, 2H, H–N(NH2)), 6.68 (d, J = 6.8 Hz, 2H, H–C(3′,5′)), 7.32 (t, J = 6.2 Hz, 1H, H–C(5)), 7.46 (bd, J = 5.6 Hz, 2H, H–C(4,6)), 7.51 (d, J = 12.5 Hz, 1H, H–Cα(vinyl)), 7.60 (s, 1H, H–C(2)), 7.68 (d, J = 12.5 Hz, 1H, H–Cβ(vinyl)), 7.91 (d, J = 6.8 Hz, 2H, H–C(2′,6′)). HRMS: Calcd [(M+) = C15H12NOCl35] 257.0608; found 257.0595 m/z (45.8%), calcd [(M+) = C15H12NOCl37] 259.0567; found 259.0575 m/z (15.5%).
(E)-1-(4′-Aminophenyl)-3-(4-chlorophenyl)- 2-propen-1-one (27)
Compound 27 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5 mmol) and 4-chlorobenzaldehyde (0.652 g, 5 mmol). Yield 84%. m.p. 69–70 °C. IR (KBr): ν = 3454, 3350, 3245, 1656, 1581, 1556, 842. 1H NMR (400 MHz, CDCl3): δ = 4.14 (bs, 2H, H–N(NH2)), 6.68 (d, J = 6.6 Hz, 2H, H–C(3′,5′)), 7.36 (d, J = 6.6 Hz, 1H, H–C(6)), 7.43 (d, J = 6.5 Hz, 1H, H–C(5)), 7.51 (d, J = 15.6 Hz, 1H, H–Cα(vinyl)), 7.55 (d, J = 15.6 Hz, 1H, H–Cβ(vinyl)), 7.81 (d, J = 6.5 Hz, 1H, H–C(3)), 8.02 (d, J = 6.6 Hz, 1H, H–C(2)), 8.12 (d, J = 6.6 Hz, 2H, H–C(2′,6′)). HRMS: Calcd [(M+ ) = C15H12NOCl35] 257.0608; found 257.0588 m/z (100%), Calcd [(M+) = C15H12NOCl37] 259.0567; found 259.0559 m/z (32.5%).
(E)-1-(4′-Aminophenyl)-3-(2-bromophenyl)- 2-propen-1-one (28)
Compound 28 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5 mmol) and 2-bromobenzaldehyde (0.584 mL, 5 mmol). Yield 83%. m.p. 127–128 °C. IR (KBr): ν = 3333, 3228, 1651, 1597, 1584, 752. 1H NMR (400 MHz, CDCl3): δ = 4.76 (bs, 2H, H–N(NH2)), 6.68 (d, J = 8.2 Hz, 2H, H–C(3′,5′)), 7.20 (t, J = 7.0 Hz, 1H, H–C(4)), 7.32 (bt, J = 6.8 Hz, 7.8 Hz, 1H, H–C(5)), 7.41 (d, J = 15.5 Hz, 1H, H–Cα(vinyl)), 7.62 (d, J = 7.8 Hz, 1H, H–C(6)), 7.69 (d, J = 7.1 Hz, 1H, H–C(3)), 7.90 (d, J = 8.2 Hz, 2H, H–C(2′,6′)), 8.04 (d, J = 15.5 Hz, 1H, H–Cβ(vinyl)). HRMS: Calcd [(M+) = C15H12NOBr79] 301.0103; found 301.0087 m/z (40.5%), Calcd [(M+) = C15H12NOBr81] 303.0082; found 303.0072 m/z (41.3%).
(E)-1-(4′-Aminophenyl)-3-(4-methylphenyl)- 2-propen-1-one (29)
Compound 29 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5 mmol) and 4-methylbenzaldehyde (0.589 mL, 5 mmol). Yield 71%. m.p. 219–220 °C. IR (KBr): ν = 2910, 2862, 2575, 1650, 1600, 1597. 1H NMR (400 MHz, CDCl3): δ = 4.51 (bs, 2H, H–N(NH2)), 7.04 (d, J = 8.7 Hz, 2H, H–C(3,5)), 7.21 (d, J = 7.7 Hz, 2H, H–C(3′,5′)), 7.41 (d, J = 15.6 Hz, 1H, H–Cα(vinyl)), 7.51 (d, J = 7.7 Hz, 2H, H–C(2′,6′)), 7.77 (d, J = 15.6 Hz, 1H, H–Cβ(vinyl)), 7.91 (d, J = 8.7 Hz, 2H, H–C(2,6)). HRMS: Calcd [(M+) = C16H15NO 237.1154]; found 237.1155 m/z (100%).
(E)-1-(2′-Hydroxyphenyl)-3-(2-hydroxyphenyl)- 2-propen-1-one (30)
Compound 30 was synthesized by the general procedure using 2′-hydroxyacetophenone (0.601 mL, 5 mmol) and 2-hydroxybenzaldehyde (0.388 mL, 5 mmol). Yield 78%. m.p. 160–162 °C. IR (KBr): ν = 3445–3450, 3060, 2915, 1632, 1570, 1265. Anal. Calcd: C, 74.99%, H, 5.03%. Found: C, 74.86%, H, 5.04%.
(E)-1-(2′,3′,4′-Trimethoxyphenyl)-3-(2-hydroxyphenyl)- 2-propen-1-one (31)
Compound 31 was synthesized by the general procedure using 2′,3′,4′-trimethoxyacetophenone (0.677 mL, 5 mmol) and 2-hydroxybenzaldehyde (0.388 mL, 5 mmol). Yield 81%. m.p. 114–116 °C. IR (KBr): ν = 3230–3450, 2986, 2967, 2874, 1634, 1595. 1H NMR (300 MHz, CDCl3): δ = 3.88, 3.92, 3.94 (s, 3H, H–C(OCH3)), 6.73 (d, J = 9.0 Hz, 1H, H–C(5′)), 6.77 (d, J = 7.8 Hz, 1H, H–C(3)), 6.91 (d, J = 7.8 Hz, 1H, H–C(5)), 6.98 (t, J = 8.4 Hz, 1H, H–C(4)), 7.25 (d, J = 16.5 Hz, 1H, H–Cα(vinyl)), 7.58 (m, 2H, H–C(6,6′)), 8.07 (d, J = 16.4 Hz, 1H, H–Cβ(vinyl)). Anal. Calcd: C, 68.78%, H, 5.77%. Found: C, 68.74%, H, 5.76%.
(E)-1-(3′,4′,5′-Trimethoxyphenyl)-3-(2-hydroxyphenyl)- 2-propen-1-one (32)
Compound 32 was synthesized by the general procedure using 3′,4′,5′-trimethoxyacetophenone (1.05 g, 5 mmol) and 2-hydroxybenzaldehyde (0.388 mL, 5 mmol). Yield 75%. Oil. IR (KBr): ν = 3400–3200, 2949, 2987, 2838, 1631, 1604. 1H NMR (300 MHz, CDCl3): δ = 3.91–3.92 (bs, 9H, H–C(OCH3)), 6.93 (t, J = 8.7 Hz, 2H, H–C(3,6)), 7.15(t, J = 7.2 Hz, 1H, H–C(4)), 7.19–7.28 (m, 3H, H–C(2′,6′,5)), 7.39 (d, J = 15.3 Hz, 1H, H–Cα(vinyl)), 7.83 (d, J = 15.4 Hz, 1H, H–Cβ(vinyl)). Anal. Calcd: C, 68.78%, H, 5.77%. Found: C, 68.75%, H, 5.76%.
(E)-1-(2′,4′-Dibromophenyl)-3-(2-hydroxyphenyl)-2-propen-1-one (33)
Compound 33 was synthesized by the general procedure using 2′,4′-dibromoacetophenone (1.39 g, 5 mmol) and 2-hydroxybenzaldehyde (0.388 mL, 5 mmol). Yield 61%. Oil. IR (KBr): ν = 3220–3430, 1659, 1562, 796. Anal. Calcd: C, 47.16%, H, 2.64%. Found: C, 47.18%, H, 2.64%.
(E)-1-(Phenyl)-3-(4-fluorophenyl)-2-propen-1-one (34)
Compound 34 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and 4-fluorobenzaldehyde (0.536 mL, 5 mmol). Yield 70%. m.p. 78–80 °C. IR (KBr): ν = 3043, 1658, 1594, 1157. 1H NMR (300 MHz, DMSO-d6): δ = 7.30 (t, J = 8.5 Hz, 2H, H–C(2)), 7.57 (t, J = 7.3 Hz, 2H, H–C(3′,5′)), 7.67 (t, J = 7.3 Hz, 1H, H–C(4′)), 7.74 (d, J = 14.6 Hz, 1H, H–Cα(vinyl)), 7.91 (d, J = 15.9 Hz, 1H, H–Cβ(vinyl)), 7.98 (dd, J = 9.0 Hz, 7.0 Hz, 2H, H–C(3,5)), 8.15 (d, J = 7.3 Hz, 2H, H–C(2′,6′)).
(E)-1-(Phenyl)-3-(3-hydroxy-4-methyoxyphenyl)- 2-propen-1-one (35)
Compound 35 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and isovanillin (0.760 g, 5 mmol). Yield 78%. m.p. 108–110 °C. IR (KBr): ν = 3400–3200, 3057, 2913, 1632, 1604. 1H NMR (300 MHz, CDCl3): δ = 3.98 (s, 3H, H–C(OCH3)), 6.97 (d, J = 8.1 Hz, 1H, H–C(5)), 7.15 (d, J = 1.8 Hz, 1H, H–C(2)), 7.23 (dd, J = 8.1 Hz, 1.8 Hz, 1H, H–C(6)), 7.40 (d, J = 15.9 Hz, 1H, H–Cα(vinyl)), 7.52 (t, J = 7.8 Hz, 2H, H–C(3′,5′)), 7.60 (t, J = 7.2 Hz, 1H, H–C(4′)), 7.77 (d, J = 15.3 Hz, 1H, H–Cβ(vinyl)), 8.02 (d, J = 8.4 Hz, 2H, H–C(2′,6′)).
(E)-1-(Phenyl)-3-(4-N,N-dimethylaminophenyl)- 2-propen-1-one (36)
Compound 36 was synthesized by the general procedure using acetophenone (0.584 mL, 5 mmol) and 4-N,N-dimethylaminobenzaldehyde (0.745 g, 5 mmol). Yield 65%. m.p. 105–106 °C. IR (KBr): ν = 2904, 1657, 1584. 1H NMR (400 MHz, CD3OD): δ = 3.03 (s, 6H, H–C(N(CH3)2)), 6.75 (dd, J = 7.3 Hz, 1.7 Hz, 2H, H–C(3,5)), 7.21 (dd, J = 7.8 Hz, 1.6 Hz, 2H, H–C(2,6)), 7.46 (d, J = 15.5 Hz, 1H, H–Cα(vinyl)), 7.52 (ddd, J = 8.4 Hz, 7.8 Hz, 1.4 Hz, 2H, H–C(3′,5′)), 7.55–7.56 (m, 2H, H–C(2′,6′)), 7.74 (d, J = 15.4 Hz, 1H, H–Cβ(vinyl)), 8.01–8.02 (m, 1H, H–C(4′)). Anal. Calcd: C, 81.24%, H, 6.82%, N, 5.57%. Found: C, 81.48%, H, 6.98%, N, 5.59%.
(E)-1-(4′-Aminophenyl)-3-(3-bromophenyl)- 2-propen-1-one (37)
Compound 37 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5 mmol) and 3-bromobenzaldehyde (0.580 mL, 5 mmol). Yield 85%. m.p. 155–157 °C. IR (KBr): ν = 3334, 3215, 1649, 1598, 1582, 749. 1H NMR (400 MHz, CDCl3): δ = 4.13 (bs, 2H, H–N(NH2)), 6.62 (d, J = 8.2 Hz, 1H, H–C(6)), 6.68 (d, J = 8.3 Hz, 2H, H–C(3′,5′)), 7.40 (d, J = 15.8 Hz, 1H, H–Cα(vinyl)), 7.65 (s, 1H, H–C(2)), 7.66 (d, J = 15.8 Hz, 1H, H–Cβ(vinyl)), 7.91 (d, J = 8.1 Hz, 1H, H–C(4)), 7.94 (d, J = 8.3 Hz, 2H, H–C(2′,6′)), 8.09 (t, J = 8.2 Hz, 8.1 Hz, 1H, H–C(5)).
(E)-1-(4′-Aminophenyl)-3 -(4-N,N-dimethylaminophenyl)- 2-propen-1-one (38)
Compound 38 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5 mmol) and 4-N,N-dimethylaminobenzaldehyde (0.744 g, 5 mmol). Yield 75%. m.p. 140 °C. IR (KBr): ν = 3332, 3226, 2904, 1657, 1584. 1H NMR (400 MHz, CDCl3): δ = 2.48 (s, 6H, H–C(N(CH3)2)), 4.09 (s, 2H, H–N(NH2)), 6.62 (bd, J = 8.1 Hz, 4H, H–C(3,5,3′,5′)), 7.14 (d, J = 13.0 Hz, 1H, H–Cα(vinyl)), 7.78 (bd, J = 8.1 Hz, 4H, H–C(2,6,2′,6′)), 7.90 (d, J = 13.0 Hz, 1H, H–Cβ(vinyl)).
Cytotoxicity studies
Brine shrimp was used as test animal for investigation of cytotoxicity according to the standard reported procedure (Ansari et al., Citation2005; Atta-ur-Rahman et al., Citation2001). Artificial saline water was prepared with 30–35 ppm saline (3.8 g sea salt/L) and placed in a small unequally divided tank. About one teaspoon of brine shrimp eggs were added to the larger compartment covered with aluminum foil. Eggs were maintained under strong aeration at room temperature for two days so that napulii were mature enough to test. The illuminated compartment attracted phototropic napulii through perforations in the dam.
Different concentrations (1000, 100 and 10 µg/mL) of the test samples were prepared in DMSO. These test samples were taken into separate test tubes, so that each test tube contained not more than 50 µL of DMSO. Subsequently, 10 brine shrimps were transferred to each test tube using micro pipettes. All tests were performed in replicates at each concentration. The vials were maintained under strong illumination. After 24 h elapse, the number of survived napulii were counted and recorded with the aid of a 3× magnifying glass. The percentage of lethality of brine shrimp nauplii was counted at each concentration for each sample. An approximate linear correlation was observed when the logarithm of concentration was plotted against the percentage of mortality and the values of LC50 (95% confidence intervals) were calculated using a simple PC program; Probid analysis. DMSO was used as a negative control.
CoMFA analysis
Data set for analysis
The data set comprised a total of 33 chalcones, whose cytotoxic potential was evaluated with the help of BSL assay. Log LC50 values were used as a dependent variable in the QSAR study. The data set was divided in two sub-sets: the training set and the test set. The training set comprised 24 chalcones (1, 3–8, 10, 12–14, 16–18, 20–21, 24, 26–28, 30–33) while the test set comprised remaining nine chalcones (2, 9, 11, 15, 19, 22–23, 25 and 29).
Computer modeling
All molecular modeling studies were performed on a Silicon graphics workstation using the molecular modeling package SYBYL 7.0 (2004), Tripos Inc. (St. Louis, MO). The structures of all 35 compounds were generated by Guassian View, Guassian Inc. (Wallingford, CT) and minimized with the Austin Model 1 (AMI) parameterization. Molecular conformation and alignment was done for carrying out CoMFA analysis.
Molecular conformation
Tripos force field, conjugated gradient method, subsequently Gasteiger–Hückel partial charges were calculated for all the chalcone analogs (Gasteiger & Marsili, Citation1980). Afterwards, the geometries of the molecules were optimized keeping a minimum gradient difference of 0.001 kcal/mol Å as a convergence criterion. The lowest energy conformation found for each molecule in the data set led to an optimization by means of HF/3–21 g* level using Gaussian 94 (Dobbs & Hehre, Citation1987).
Structural alignment
This is perhaps the most subjective and critical step in CoMFA study, in as much as the resulting 3D QSAR model is often sensitive to the particular alignment scheme (Liu et al., Citation2003). Since the specific molecular target of these compounds in brine shrimps is unknown, the docking-based alignment method for developing a QSAR model could not be used. Instead, an active molecule in the training set, compound 5 (), was chosen as the template and the data set was superposed on the template conformation with the application of the database alignment method. Out of the three alignments of superimposition, i.e., rms fitting, flexible fitting and rigid body field fit, rms fitting was adopted. Each molecule from the training set was superimposed to the template by minimizing the rms distances between each pair of corresponding atoms of the template and the compound to be aligned. All structures were fully geometry-optimized using standard Tripos force field with a distance-dependent dielectric function until a root mean square (rms) deviation of 0.001 kcal/mol/Å was achieved (Clark et al., Citation1989). The common fitting centers selected for aligning all chalcones are shown by asterisk ().
Figure 1. Fitting centers used for overlapping chalcones.
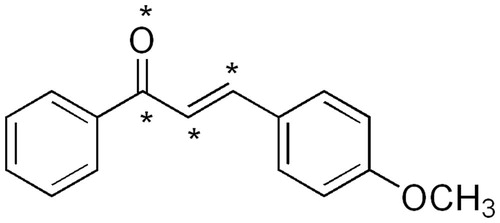
Table 1. The brine shrimp cytotoxicity of the synthesized chalcones.  .
.
Antitumor activity
Human tumor cell lines
Human lung carcinoma (H1299), squamous cell carcinoma (H157), malignant melanoma of skin (HT144), breast cancer (MCF7), colon cancer (SKCO1) and liver cancer (HepG2) cell lines obtained from American Type Culture Collection (Manassas, VA) were used to check the cytotoxicity of the five synthesized chalcone derivatives. All cell lines were grown at 37 °C, 5% CO2 in the RPMI 1640 medium or DMEM as per the requirement of each cell line.
Cell proliferation assay
Cell lines were grown in 96-well tissue culture grade plates in appropriate concentrations. In vitro Sulforhodamine B assay and Trypan blue exclusion tests were carried out to assess the cytotoxicity of compounds (34–38) (Keepers et al., Citation1991) using different concentrations of each compound. Briefly, cells were seeded on 96-well micro titer plates 24 h before the addition of compounds to allow them to form a monolayer. This was followed by the addition of the synthesized compounds using methotrexate and vincristine as controls. The cells were incubated at 37 °C for additional 24 h. The cells were then fixed for the detection of cell viability and absorbance was taken at 560 nm with Platos R496 Microplate reader, AMP Diagnostics (Graz, Austria). IC50 values were calculated using a nonlinear regression analysis.
Tubulin polymerization assay
In vitro tubulin polymerization-depolymerization efficacy of chalcones was assessed through a turbidity based tubulin polymerization measured at 340 nm according to the reported protocol (Huang et al., Citation2005; Lee & Timasheff, Citation1977; Tuma et al., Citation2010). Lyophilized bovine tubulin HTS02, Cytoskeleton, Inc. (Denver, CO) was suspended in G-PEM buffer (80 mM 1,4-piperazinediethane sulfonic acid pH 7, 1 mM EGTA, 1 mM MgCl2, 1 mM GTP, 5% glycerol) to a final concentration of 3 mg/mL and kept at 4 °C. The test compounds 34–38, taxol and colchicine were prepared as 100 × DMSO stock solutions. Compounds (10 μL) were added to the taxol and colchicine into a pre-warmed 96-well flat bottom plate. The plates were placed back into warm plate reader at 37 °C for 1 min. 100 µL of the tubulin reaction mix was added into each of the eight wells using a single channel pipettor. Plate contents were mixed by shaking for 5 s, and absorbance was measured at 1 min intervals for 60 min using a PowerWave HT (Winooski, VT) microplate reader.
Results and discussion
BSL studies
A parallel library of 33 differently substituted chalcones was synthesized under microwave-assisted irradiation through the Claisen–Schmidt condensation protocol. The members of the library were confirmed through physical data and spectroscopic analysis: Infra-red spectroscopy (IR), proton nuclear magnetic resonance spectroscopy (1H NMR), high-resolution mass spectroscopy (HRMS) and elemental analysis. These compounds were evaluated for their cytotoxic potential through BSL assay (Anderson et al., Citation1991; Manilal et al., Citation2009). BSL cytotoxicity data of these chalcones was collected in the form of LC50 values and the potencies were found within a range of 0.1 to ∼834 μM. describes the structures of all these compounds and LC50 values obtained. The activity data of this diverse set of chalcone analogs was used for the development and validation of CoMFA model.
CoMFA analysis
CoMFA interaction energies
The standard Tripos settings were used to carry out the CoMFA analysis. To derive the CoMFA fields, a 3D cubic lattice was created; the steric and electrostatic parameters were calculated at each lattice intersection of regularly spaced grid of 2.0 Å in all three dimensions within the defined region. The aligned molecules were placed in a 3D grid box such that the entire set was included in it. The van der Waals potential and the Coulombic term representing the steric and electrostatic fields were calculated using standard Tripos force fields. An sp3 carbon atom with a positive charge (+1) was used as a probe atom to generate steric (Lennarde Jones potential) field energies and electrostatic (Coulombic potential) field energies. A distance-dependent dielectric constant of 1.00 was used. The steric and electrostatic fields were truncated to +30.00 kcal/mol. The CoMFA fields generated automatically were scaled by the CoMFA-STD method in SYBYL.
PLS analysis
The CoMFA descriptors were used as independent variables and inverse logarithm of LC50 values (pLC50) as a dependent variable in PLS regression analysis for deducing the 3D-QSAR model. Normally, cross-validation is employed to check the predictivity of the derived model. The performance of models was calculated using the leave-one-out (LOO) cross-validation method. The optimum number of components used to derive the non-cross-validated model was defined as the number of components leading to highest r2 cross-validated and lowest standard error of prediction (SEP). To speed up the analysis and reduce noise, a minimum filter value of 2.00 kcal/mol was used. Only those steric and electrostatic energies with values ≥2.0 kcal/mol were considered in the PLS linear regression analysis and the predictive quality of the “best” correlation model was determined. PLS analysis was used to correlate the biological activity index with CoMFA values that contained the magnitude of either the steric or electronic potential (Clark et al., Citation1990). CoMFA model was obtained with 24 chalcones in the training set and nine chalcones in the test set, respectively. The actual, predicted and residual values of all the training and test set compounds are shown in and , respectively.
Table 2. Observed and predicted pLC50 of the training set compounds.
Table 3. Observed and predicted pLC50 of the test set compounds.
Cross-validation runs with varying number of groups were also performed to improve the confidence limits of the derived model. In the best model, when performed with the optimal number of five components and no validation PLS analysis, q2 and r2 values were obtained as 0.626 and 0.950, respectively, whereas the F value was 100.754 and the Standard Error Estimate (SEE) was found to be 0.335. The plots of observed versus predicted cytotoxicity of training and test set chalcones are presented as and . The CoMFA electrostatic and steric fields based on PLS analysis are presented as 3D contour plots in . The contribution of steric and electrostatic field is 31.1% and 68.9%, respectively.
Figure 2. Calculated versus observed activity of the training set compounds.
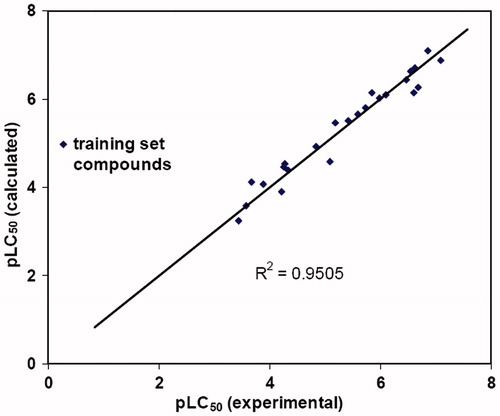
Figure 3. Calculated versus observed activity of the test set compounds.
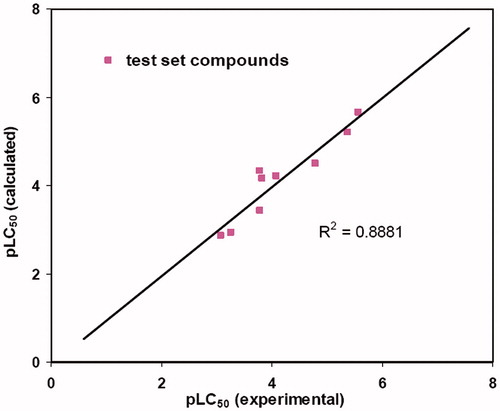
Figure 4. Stereoview of the CoMFA map for the electronic and steric contributions favoring activity.
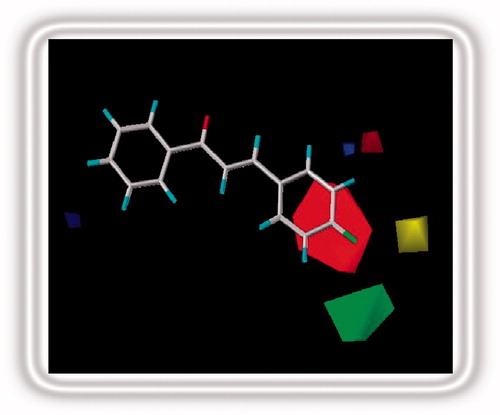
As is evident in and , there is no outlier in the CoMFA model and the largest residual found in chalcone 28 is 0.49 falls within ±0.5 log unit. Furthermore, the r2 value of the test set compounds is highly indicative of a competent QSAR model derived from the training set compounds. In the CoMFA method, results are presented as contour maps that correlate the change in biological activity with the molecular field values. The steric contour maps are represented in green and yellow colors while the electrostatic contours are depicted in red and blue colors. Red color indicates areas where more negative density favors activity and blue color indicates areas where more positive density promotes the activity. Green color indicates areas where bulky substituents favor activity and yellow color indicates areas where less bulky substituents promote activity. The contour maps are limited to the regions where various substituents have been modified and have an impact upon activity of resulting analogues.
In silico designing
Based on the 3D-QSAR analysis, predictions were made for favorable electronic and steric regions that are expected to enhance activity in the resulting chalcone derivatives. These results are shown in , which gives an appropriate description of the favorable electrostatic and steric substitution pattern.
Figure 5. Favorable electronic and steric regions for enhanced potency in cytotoxic chalcones.
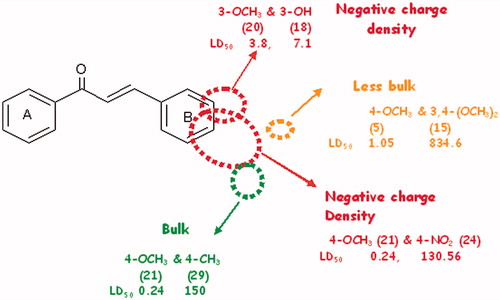
It may be generalized that chalcones bearing donors at 3 and 4 positions are expected to result in increased cytotoxic potential. Furthermore, bulk at 4′ position seems favorable for activity whereas bulk at 3 and 4 positions may result in steric conflict leading to decreased potential of the chalcone derivatives. The figure thus presented provides an insight for designing more chalcone derivatives that may result in enhanced activity.
Based on favorable steric and electrostatic regions obtained from CoMFA analysis, some new compounds were designed with an expectation of improved cytotoxic potential. Each of the synthesized compounds retained one or more electrostatic or steric requirement, predicted as essential for activity in CoMFA studies. A large red isopleth at position 4 of ring B indicates that electron density at this position strongly favors the cytotoxic activity. Strong electron donor, e.g., N(CH3)2 was, therefore, introduced in the designed chalcones. Furthermore, green and yellow isopleths at position 4 indicated that both bulky and small functional groups are favorable for activity. Therefore, fluoro group being small in size and having low polarizability donates electron density to phenyl ring, making position 4 of ring B as electron rich. This derivatization favors both electrostatic and steric requirement and is expected to enhance activity. The 3- or 4-methoxy substituted chalcones resulted in good activities, whereas activity dropped drastically when the 3,4-dimethoxy substituted ring B was present. The position of yellow isopleth indicated that bulk both at 3 and 4 positions is sterically unfavorable. One of two methoxy substituent at 3 and 4 positions of ring B (chalcone 15) was replaced by a less bulky OH group. Furthermore, an electron donor, e.g., –Br was added at position 3 of ring B, which was expected to result in fair activity. As a result, a total of five compounds (34–38) were designed and their pLC50 values were predicted from the derived QSAR equation based on CoMFA studies (). On the basis of calculated pLC50 values, compound 34 was expected to be the most potent, whereas compound 37 as the least potent.
Table 4. Predicted pLC50 of the designed chalcones.
Molecular docking
The cytotoxic potential of designed chalcones was further evaluated through their docking into tubulin protein. Tubulin polymerizes and assembles in the cellular environment into microtubules (MTs) (Mozziconacci et al., Citation2008). Stabilization of MT dynamics is a common mechanism of antimitotic agents resulting in apoptosis of cancer cells (Jordan & Wilson, Citation1998). MTs and tubulin dimers are dynamic targets for cancer chemotherapy. Tubulin crystal structures are indispensable to determine the mechanism of the action of different antitumors which are known to target tubulin. The tubulin-to-tubulin-binder interactions obtained through X-ray crystal structure data provide a valuable tool for in silico studies of chalcones interactions with tubulin protein, thus providing a new arsenal for structure-based drug design (Aldaz et al., Citation2005). In view of the significance of tubulin as a target of anticancer drugs, it was selected for conducting molecular docking studies on the cytotoxic chalcones, identified as a result of the BSL and CoMFA assay. Chalcones, as already stated, are known to bind to tubulin dimer halting mitosis, resulting in their antimitotic action and subsequent apoptosis in cancer cells (Liu & Go, Citation2006). For tracing binding interactions and conformations of chalcone as tubulin binders, the docking studies of all the chalcones were performed using MOE software.
Docking procedure
The crystal structure of tubulin protein complex was obtained from the Protein data bank (PDB id: 1z5v) complexed with molecular ligand GSP (guanosinediphosphatemonothio phosphate) (Aldaz et al., Citation2005). The X-ray crystallographic structure of the complex was used for docking calculations after ligand removal and enzyme adjustment. The water molecules and ligand were removed from the imported protein in MOE and all possible hydrogen atoms were added to the structure with their standard geometry. The backbone and the residues were kept fixed and an energy minimization was performed for all the added hydrogen atoms. The resulting model was subjected to systematic conformational search where all items were set as default with RMSD gradient of 0.01 kcal/mol using the Site Finder tool. MOE alpha Site Finder was used for the active site search in the enzyme structure and dummy atoms were created from the obtained alpha spheres. The target compounds were built using the builder interface of MOE program and subjected to energy minimization tool using the MOPAC 7.0. Chalcone derivatives were docked into the active site and interactions were studied. Ten docking conformations were selected for each ligand and the best conformation of each of the ligand–receptor complex was selected based on energy grounds. A further energy minimization was used to refine the orientation of substrate in the binding site of tubulin. The ligand–enzyme complex model obtained henceforth was then used for calculating the energy parameters using MMFF94× force field energy calculation and predicting the ligand–enzyme interaction at the active site. The docked conformations were ranked according to the interaction affinity.
Validation of docking reliability
Before using the tubulin crystal structure for docking chalcones, it was necessary to validate the docking reliability. The known X-ray crystal structure of tubulin obtained from Protein data bank (PDB id: 1z5v) was found complexed with molecular ligand GSP and was confirmed by the redocking method. The RMSD value was used as a measure of how close the predicted structure of GSP was bound to receptor. As expected, the position and orientation of predicted ligand and redocked ligand were well overlapped (RMSD-tolerance value computed using SVL program was 1 Å). Therefore, the docking parameters used for GSP were extended for searching the binding conformation of chalcones to the active site.
Docking results
The designed inhibitors were docked into the binding site of tubulin. Binding free energies were calculated using the MMFFX94 free energy calculation method (Basili et al., Citation2007). Free energy of inhibitor binding, Gbind, was obtained from the difference between the free energy of the receptor–ligand complex (Gcpx) and the unbound receptor (Grec) and ligand (Glig) according to the following equation.
Energy scores of the designed chalcones are shown in . Designed chalcone, 34, was predicted as the most active from the CoMFA model, it was also found as the most potent in docking studies. Descending order of the Gibbs free energy of binding was found to be the same as the descending order of predicted activities from the CoMFA model. In the binding pocket, compounds 34–38 showed weak hydrogen bonding and hydrophobic interactions. Chalcone 34 was found to show 50% H-bonding with Ser 140 whereas hydrophobic interactions with Phe 225. Chalcone 35 showed 36% hydrogen bonding with Asn 207 whereas 36 showed 40% to the same. Chalcone 37 showed hydrophobic interactions with Phe 225 and 38 showed 15% hydrogen bonding to Asp 180. The binding pose of the most potent analog 34 is presented in .
Figure 6. Chalcone 34 bound into the binding pocket of tubulin active site.
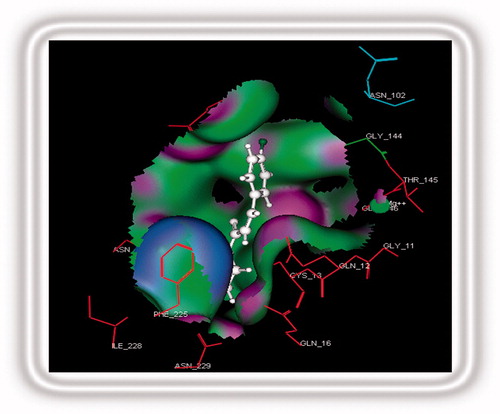
Table 5. The energy scores (kCal mol−1) of designed inhibitors.
BSL activity
The designed compounds 34–38 were synthesized and evaluated for their BSL activity for drawing any possible relationship, if exists, between the predicted cytotoxic potential of these compounds and their actual experimental values (). The observed LC50 values were recorded and compared with the predicted values from CoMFA analysis. It is readily observed from that potencies of compounds are in accordance with those predicted through the QSAR model and docking studies. The error between the actual and predicted values is also tolerable, except for the –Br substituted analog 37. Moreover, the observed potencies of designed chalcones 34–38 decrease in the same order as the predicted potencies resulted from the CoMFA model and the binding affinities portrayed through molecular docking studies. The close correlation among the predicted and observed activities of newly synthesized compounds confirms the validity of BSL assay in QSAR-based in silico designing of active analogs.
Table 6. The predicted and observed pLC50 values of the designed chalcone analogues.
Antitumor activity
Cell proliferation assay
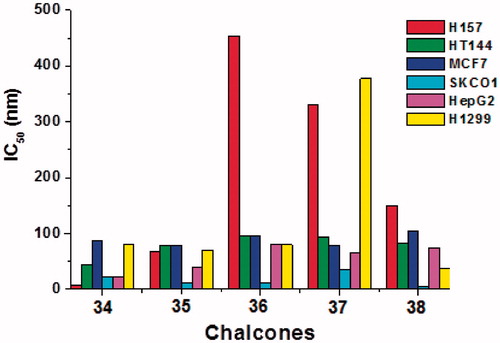
The designed compounds 34–38 were subjected to cell proliferation assay against six human tumor cell lines. The tested compounds resulted in high potencies with an IC50 values ranging from 10 to 150 µM, with only a few exceptions reaching to 450 µM. A general trend regarding the antitumor potencies is the same as predicted from their BSL and docking studies (). The potencies of the new compounds increase with decrease in binding energies of designed compounds as was calculated during chalcone–tubulin docking studies.
Figure 7. Cytotoxicity of chalcones 34–38 against human squamous cell carcinoma (H157), malignant melanoma of skin (HT144), breast cancer (MCF7), colon cancer (SKCO1), liver cancer (HepG2) and lung carcinoma (H1299).
Tubulin polymerization assay
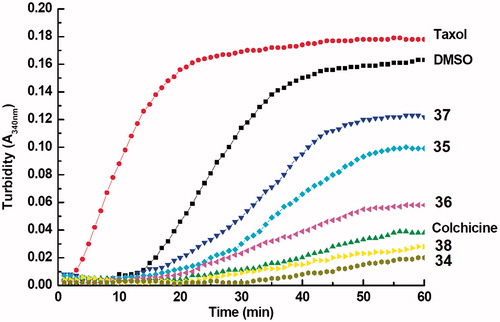
MTs pull the chromosomes apart during mitosis helping in cell division. Most of the antimitotic agents interact with the tubulin and disturb the rates of tubulin polymerization and depolymerization. The tubulin polymerization activity was therefore estimated for compounds 34–38 through a conventional absorbance based assay at 340 nm (). DMSO, taxol and colchicine were used as the positive controls. All the designed analogues and colchicine significantly inhibited tubulin polymerization at a conc. of 5 µM. Compounds 34 and 38 with 4-F and 4-N(CH3)2 substitution at ring B were found potent enough to supersede polymerization inhibition even of colchicine. The assay clearly showed the tubulin polymerization inhibition properties of the tested compounds, conferring the tubulin polymerization inhibition as the possible mechanism for such high in vivo antitumor activities of the designed compounds 34–38.
Figure 8. Tubulin polymerization activity of 5 μM of each of designed chalcones 34–38 on tubulin polymerization. Taxol and colchicine (5 μM) used as reference compounds whereas DMSO was used as control. The activity was assessed by turbidity change using UV-VIS spectrophotometry.
From the above discussion, it is evident that the CoMFA model provided a means for in silico designing of new chalcones as cytotoxic agents and an excellent agreement was observed between the predicted and observed activities of the synthesized chalcones. Molecular docking studies strengthened the agreement developed based on QSAR study. The designed compounds were found to be promising antitumor agents and tubulin polymerization inhibitors when tested on six human tumor cell lines. Furthermore, QSAR and docking studies based on the cytotoxicity to the brine shrimps confirmed the validity of the said assay as a pre-screening method for evaluating antitumor potential of any set of synthesized compounds.
Conclusion
The results of the study showed that the BSL assay may help in the prescreening, designing and synthesis of potent antitumor drugs. The 3DQSAR-based prediction of antitumor activities of derivatives 34–38 were directed by the BSL assay, leading to quite promising antitumor activities as displayed in . The predicted and the observed cytotoxicity values were also quite close with minimal errors. Furthermore, the antitumor activity of the stated group of compounds was proved to be as a consequent of their tubulin-binding properties. The tubulin-polymerization assay proposed that the tubulin-polymerization inhibition was leading to the antitumor potential of tested compounds. Keeping in view all the results of this study, we conclude that the BSL assay is an effective antitumor prescreen and may be used for reducing the cost for preliminary testing in antitumor drug discovery expeditions.
Declaration of interest
The authors declared no conflict of interest. Dr. Samina Nazir and Dr. Farzana L. Ansari are thankful for financial support from the Higher Education Commission (HEC) and Pakistan Science Foundation (PSF) Pakistan.
Acknowledgements
Dr. Zaheer-ul-Haq acknowledges Prof. Dr. Bernd M. Rode (Department of Theoretical Chemistry, University of Innsbruck, Austria) in establishing facilities for computational studies at Dr. Panjwani Centre for Molecular Medicines and Drug Research (PCMD) Karachi, Pakistan.
References
- Aryapour H, Riazi GH, Ahmadian S, et al. (2012). Induction of apoptosis through tubulin inhibition in human cancer cells by new chromene based chalcones. Pharm Biol 50:1551–60
- Ahsanullah Ansari FL, Nazir S, et al. (2007). Combinatorial synthesis and antitumour studies on an aryl and heteroaryl chalcone library using positional scanning method. Chem Biodiv 4:203–14
- Aldaz H, Rice LM, Stearns T, Agard DA. (2005). Insights into microtubule nucleation from the crystal structure of human gamma-tubulin. Nature 435:523–7
- Anderson JE, Goetz CM, McLaughlin JL, Suffness M. (1991). A blind comparison of simple bench-top bioassays and human tumour cell cytotoxicities as antitumor prescreens. Phytochem Anal 2:107–11
- Ansari FL, Nazir S, Naureen H, Mirza B. (2005). Combinatorial synthesis and antibacterial studies on an indexed chalcone library. Chem Biodivers 2:1656–64
- Ansari FL, Baseer M, Iftikhar F, et al. (2009). Microwave assisted synthesis, antibacterial activity against Bordetellabronchiseptica of a library of 3′-hydroxyaryl and heteroaryl chalcones and molecular descriptors-based SAR. ARKIVOC X:318–32
- Atta-ur-Rahman Choudhary MI, Thomsen WJ. (2001). Bioassay Techniques for Drug Development. Amsterdam, The Netherlands: Harwood Academic Publishers
- Basili S, Bergen A, DallAcqua F, et al. (2007). Relationship between the structure and the DNA binding properties of diazoniapolycyclic duplex- and triplex-DNA binders: Efficiency, selectivity, and binding mode. Biochem US 46:12721–36
- Boeck P, Falcào CAB, Leal PC, et al. (2006). Synthesis of chalcone analogues with increased antileishmanial activity. Bioorg Med Chem 14:1538–45
- Bowen JP, Robinson TP, Ehlers T, et al. (2005). Synthesis and biological evaluation of aromatic enones related to curcumin. US Patent 6,906,105
- Cai SX, Reddy PS, Drewe JA, et al. (2007). Multifluoro-substituted chalcones and analogs as activators of caspases and inducers of apoptosis and the use thereof. US Patent 72,56,219
- Clark M, Cramer RD III, Jones DM, et al. (1990). Comparative molecular field analysis (CoMFA). 2. Toward its use with 3D structural databases. Tetrahedron Comput Meth 3:47–59
- Clark M. Crammer RD III, Opdenbosh V. (1989). Validation of the general purpose Tripos 5.2 force field. J Comput Chem 10:982–1012
- Damazio RG, Zanatta AP, Cazarolli LH, et al. (2010). Antihyperglycemic activity of naphthylchalcones. Eur J Med Chem 45:1332–7
- Deng J, Sanchez T, Al-Mawsawi LQ, et al. (2007). Synthesis and biological activities of aryl-ether-, biaryl-, and fluorene-aspartic acid and diaminopropionic acid analogs as potent inhibitors of the high-affinity glutamate transporter EAAT-2. Bioorg Med Chem 15:4985–8
- Dobbs K, Hehre WJ. (1987). Molecular orbital theory of the properties of inorganic and organometallic compounds. Comput Chem 8:880–93
- Dominguez JN, Leon C, Rodrigues J, et al. (2005a). Synthesis and evaluation of new antimalarial phenylurenyl chalcone derivatives. Med Chem 48:3654–8
- Dominguez JN, León C, Rodrigues J, et al. (2005b). Synthesis and antimalarial activity of sulfonamide chalcone derivatives: II. Farmaco 60:307–21
- Ducki S. (2009). Antimitotic chalcones and related compounds as inhibitors of tubulin assembly. Anticancer Agents Med Chem 9:336–47
- Dyrager C, Wickstrom M, Friden SM, et al. (2011). Inhibitors and promoters of tubulin polymerization: Synthesis and biological evaluation of chalcones and related dienones as potential anticancer agents. Bioorg Med Chem 19:2659–65
- Furusawa J, Funakoshi TM, Mashino T, et al. (2009). Chalcones from Myracrodruonurundeuva are efficacious in guinea pig ovalbumin-induced allergic conjunctivitis. Int J Immunopharmacol 9:499–507
- Gasteiger J, Marsili M. (1980). Iterative partial equalization of orbital electronegativity – A rapid access to atomic charges. Tetrahedron 36:3219–28
- Geyer JA, Keenan SM, Woodard CL, et al. (2009). Selective inhibition of Pfmrk, a Plasmodium falciparum CDK, by antimalarial 1,3-diaryl-2-propenones. Bioorg Med Chem Lett 19:1982–5
- Gacche RN, Dhole NA, Kamble SG, Bandgar BP. (2008). In vitro evaluation of selected chalcones for antioxidant activity. J Enzyme Inhib Med Chem 23:28–31
- Ghosh A, Mandal S, Banerji A, et al. (2009). A new chalcone from Pongamiapinnata and its antioxidant properties. Nat Prod Commun 4:209–10
- Huang YT, Huang DM, Guh JH, et al. (2005). CIL-102 interacts with microtubule polymerization and causes mitotic arrest following apoptosis in the human prostate cancer PC-3 cell line. J Biol Chem 280:2771–9
- Jordan MA, Wilson L. (1998). Microtubules and actin filaments: Dynamic targets for cancer chemotherapy. Curr Option Cell Biol 10:123–30
- Kayser O, Kiderlen AF. (2001). In vitro leishmanicidal activity of naturally occurring chalcones. Phytother Res 15:148–52
- Keepers YP, Pizao PE, Peters GJ, et al. (1991). Comparison of the sulforhodamine B protein and tetrazolium (MTT) assays for in vitro chemosensitivity testing. Eur J Cancer 27:897–900
- Kiat TS, Pippen R, Yusof R, et al. (2006). Inhibitory activity of cyclohexenyl chalcone derivatives and flavonoids of fingerroot, Boesenbergia rotunda (L.), towards dengue-2 virus NS3 protease. Bioorg Med Chem Lett 16:3337–40
- Kong Y, Wang K, Edler MC, et al. (2010). A boronic acid chalcone analog of combretastatin A-4 as a potent antiproliferation agent. Bioorg Med Chem 18:971–7
- Kim MY, Park BY, Kim KM, et al. (2003). Pharmaceutical composition containing chalcone or its derivatives for matrix metalloproteinase. Patent WO/2003/037315
- Lee JC, Timasheff SN. (1977). Tubulin polymerization assay using >99% pure tubulin, OD based – Porcine (BK006P) N. Biochemistry 16:1754–62
- Lin YM, Zhou Y, Flavin MT, et al. (2002). Chalcones and flavonoids as anti-tuberculosis agents. Bioorg Med Chem 10:2795–802
- Liu X, Go ML. (2006). Antiproliferative properties of piperidinylchalcones. Bioorg Med Chem 14:153–63
- Liu M, Wilairat P, Croft SL, et al. (2003). Structure--activity relationships of antileishmanial and antimalarial chalcones. Bioorg Med Chem 11:2729–38
- Manilal A, Sujith S, Kiran S, et al. (2009). Cytotoxic potentials of red alga, Laurencia brandenii collected from the Indian coast. Global J Pharmacol 3:90–4
- Mozziconacci J, Sandblad L, Wachsmuth M, et al. (2008). Tubulin dimers oligomerize before their incorporation into microtubules. PLoS ONE 3:e3821 . doi:10.1371/journal.pone.0003821
- Na Y, Cha J, Yoon H, Kwon Y. (2009). A concise synthesis of licochalcone E and its regio-isomer, licochalcone. Chem Pharm Bull 57:607–9
- Sivakumar PM, Priya S, Doble M. (2009). Synthesis, biological evaluation, mechanism of action and quantitative structure–activity relationship studies of chalcones as antibacterial agents. Chem Biol Drug Dis 73:403–15
- Sivakumar PM, Seenivasan SP, Kumar V, Doble M. (2007). Synthesis, antimycobacterial activity evaluation, and QSAR studies of chalcone derivatives. Bioorg Med Chem Lett 17:1695–700
- SYBYL. (2004). Ligand-Based Design Manual, version 7.0. TriposInc, St. Louis (MO): SYBYL, Tripos Inc
- Tuma, AMC, Malikzay A, Ouyang X, et al. (2010). Antitumor activity of IMC-038525, a novel oral tubulin polymerization inhibitor. Translational Oncol 3:318–25
- Won SJ, Liu CT, Tsao LT, et al. (2005). Synthetic chalcones as potential anti-inflammatory and cancer chemopreventive agents. Eur J Med Chem 40:103–12
- Yang HM, Shin HR, Cho SH, et al. (2007). Structural requirement of chalcones for the inhibitory activity of interleukin-5. Bioorg Med Chem 15:104–11
- Zhou J, Geng G, Batist G, Wu J H. (2009). Synthesis and potential anti-prostate cancer activities of ionone-based chalcones. Bioorg Med Chem Lett 19:1183–96