Abstract
Context: The poor prognostic outcome of breast cancer is largely due to its resistance to cancer therapies. Development of therapeutic agents that can inhibit growth and induce apoptosis in breast cancer cells can help solve the problem. Emodin is an active anthraquinone that has been reported to have diverse biological effects.
Objective: In this study, the anticancer effects of emodin on growth inhibition, apoptosis induction and the expression of apoptosis-related genes in MCF-7 cells were investigated.
Materials and methods: Growth inhibition induced by emodin was investigated by the MTS assay and the colony formation assay; while emodin-induced apoptosis was determined by the COMET assay and DNA fragmentation detection. Emodin (35 μM)-induced alterations in the expression of apoptotic-related genes were detected by using real-time PCR.
Results: Emodin had significant growth inhibitory effects on MCF-7 cells with IC50 = 7.22 µg/ml (∼30 μM). It also exerted a concentration-dependant inhibitory effect on the colony-forming ability of MCF-7 cells with IC50 = 7.60 µg/ml (∼30 µM). Hallmarks of apoptosis, such as single-strand DNA breakage and DNA fragmentation, were observed in emodin-treated MCF-7 cells. The gene expression of Fas ligand (FASL) was up-regulated (p < 0.01) but those of MCL1, CCND1 and C-MYC were down-regulated (p < 0.05) in emodin-treated MCF-7 cells.
Discussion and conclusion: This study indicated that emodin could induce growth inhibition and apoptosis in MCF-7 cells through the modulation of the expression of apoptosis-related genes. The growth inhibitory effects of emodin might involve both the intrinsic and the extrinsic apoptotic pathways and cell cycle arrest.
Introduction
Breast cancer is the most common and lethal malignancy among women worldwide (Kummalue et al., Citation2007). The poor prognostic outcome of breast cancer is largely due to its resistance to current cancer therapies. Current approaches for treating human breast cancer include surgery, radiotherapy, hyperthermia, hormone replacement therapy and adjuvant chemotherapy. However, the effects of these conventional treatments are not always satisfactory. Therefore, recent research focus on breast cancer management is the development of better therapeutic agents that can inhibit growth and induce apoptosis in cancer cells. This is because the inhibition of cancer cell growth and the induction of cancer cell death are the two major therapeutic parameters in cancer treatment.
Emodin (1,3,8-trihydroxy-6-methylanthraquinone) is a biologically active anthraquinone derivative that naturally occurs in many widely used Chinese medicinal herbs, such as Rheum officinale Baill (Polygonaceae). A water extract of R. officinale has been shown by our laboratory to have anticancer effects on MCF-7 cells (Li et al., Citation2009). Many previous reports have indicated that emodin possesses diverse biological effects, such as antibacterial, diuretic, immunosuppressive, anti-inflammatory and vasorelaxant activities (Basu et al., Citation2005; Huang et al., Citation1991a,Citationb; Kim et al., Citation2005; Li et al., Citation2005; Zhang et al., Citation2005). Emodin has also been reported to have anti-proliferative effects on several human cancer cell lines (Chen et al., Citation2002; Huang et al., Citation2004, Citation2008; Lee, Citation2001; Lee et al., Citation2001; Srinivas et al., Citation2003) through different molecular mechanisms. However, the anti-proliferative effect of emodin on MCF-7 cells and the anticancer mechanisms involved have not been clearly demonstrated.
Apoptosis refers to programmed cell death, a common physiological process, to remove unwanted or dying cells from the body. Apoptosis can be induced either by the intrinsic pathway which is initiated by mitochondria or by activating the death receptor (extrinsic) and it may provide the mechanistic basis for the anticancer effects of emodin on MCF-7 cells. Cell shrinkage and mono- or oligo-chromosomal DNA breakage are some of the characteristics of apoptosis. Any modulation on the expressions of apoptosis-related genes could also trigger apoptosis. In recent years, many reports have revealed that modulating the expression level of apoptosis-related genes, such as Fas ligand (FASL), myeloid cell leukemia sequence 1 (MCL1), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), BCL2-associated X protein (BAX), cyclin D1 (CCND1) and v-myc myelocytomatosis viral oncogene homolog (C-MYC), provides therapeutic and chemoprevention targets for the treatment of cancer through the induction of cell cycle arrest and apoptosis (Ahn et al., Citation2006; Chang et al., Citation2004; Ishitani et al., Citation2003; Jang et al., Citation2004; Lee et al., Citation2005; Lin et al., Citation2005; Petty et al., Citation2003; Way et al., Citation2005; Wu et al., Citation2005; Zhang & Huang, Citation2006).
In this study, we examined the anticancer effects of emodin on cell growth inhibition, colony formation inhibition, apoptosis induction (DNA fragmentation detection and COMET assays), and the expression of apoptosis-related genes in MCF-7 cells. The results indicate that emodin could significantly inhibit cell growth and colony formation and induce apoptosis in MCF-7 cells through modulation of the expression of apoptosis-related genes.
Materials and methods
Chemicals and materials
The cell titer 96 aqueous MTS reagent was purchased from Promega Chemicals Co. (Madison, WI). All other chemicals used were of analytical grade. Cell culture medium, Dulbecco’s modified eagle medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco Laboratories (Grand Island, NY). The human breast carcinoma MCF-7 cell line (HTB-22) was purchased from the American Type Culture Collection (Manassas, VA). Emodin (purity > 98%, HPLC grade) was purchased from Sigma-Aldrich (St. Louis, MO). Emodin was dissolved in DMEM with 0.1% DMSO and stored at −20 °C until use.
Cell culture
MCF-7, the human breast carcinoma cell line, was maintained in low-glucose DMEM supplemented with 10% (v/v) FBS, 100 units/mL penicillin and 100 µg/mL streptomycin (Sigma, St. Louis, MO) in a culture plate at 37 °C in a humidified incubator with 5% carbon dioxide. The media of the cells were changed every 2–3 d and the cells were sub-cultured once they reached 70–80% confluence.
MTS assay
The inhibitory effects of emodin on cell growth were measured by MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] growth inhibition assay, which is a non-radioactive cell proliferation assay and a colorimetric method for determining the number of viable cells. The MTS growth inhibition assay was performed according to the instructions provided by the manufacturer (Promega). Briefly, 3000 cells were seeded into each well of a 96-well plate (Falcon) on day 1. On day 2, the cells were either treated with different concentrations of emodin for different incubation periods (24, 48 or 72 h) or remained as untreated controls at 37 °C with 5% carbon dioxide. At the end of each time point, fresh complete medium containing 10 µL of MTS solution was added and further incubation for 2 h was carried out. The optical density of each culture was subsequently recorded at 490 nm using a microplate reader (Bio-Rad, model 550, Hercules, CA). Each experiment was performed in triplicate. Results are expressed as the percentage of growth inhibition with respect to the untreated cells (control).
Soft agar colony formation assay
Soft agar colony formation assays were performed using a double-layer soft-agar method. In each well of a 6-well plate, 20 000 cells were mixed with different concentrations of emodin and plated in top agar (0.5% agarose) over a base agar of 0.6% agarose. Cells were incubated for 7 to 14 d under standard conditions. Colonies with more than 50 cells were counted.
COMET assay (single-cell gel electrophoresis)
Drug-induced DNA damage was analyzed using the COMET assay (Klaude et al., Citation1996; Singh et al., Citation1988) with slight modifications. Cell pellets (treated with various concentration of emodin for 72 h) were collected by centrifugation and re-suspended with 200 µL of phosphate-buffered saline (PBS) and 800 µL of 1% low-melting point (LMP) agarose. The mixture was subsequently pipetted onto frosted glass microscope slide pre-coated with a layer of 1.0% normal melting point agarose prepared in PBS. The LMP agarose covered with a cover slip and incubated at 4 °C for 10 min. After the solidification of LMP agarose, the cover slips were gently removed and replaced with another 0.8% LMP agarose pre-coated cover slip with its 0.8% LMP agarose facing the solidified LMP agarose, and allowed to solidify at 4 °C for 10 min. After 10 min, the cover slip was removed and the cells were lysed in high salt solution (2.5 M NaCl, 10 mM Tris-HCl, 100 mM EDTA, pH 10, with 1% Triton and 10% dimethyl sulfoxide) for 1 h. The slide was then placed in a horizontal electrophoresis unit with fresh buffer (1 mM EDTA, 300 mM NaOH, pH 13) and incubated for 20 min to allow unwinding of DNA. Electrophoresis was then conducted in freshly prepared electrophoresis buffer (pH 13) for 20 min at 25 V and 300 mA (0.8 V/cm) at 4 °C. Subsequently, the slides were gently washed with neutralization solution (0.4 M Tris-HCl, pH 7.5) for 20 min and stained with 20 µL ethidium bromide (15 µg/mL). Stained nucleoids were scored visually using a fluorescence microscope (Leica, Wetzlar, Germany) equipped with a digital camera. One hundred comets on two slides were acquired using the IM50 software image analysis system (Leica, Wetzlar, Germany). Tail length was calculated and expressed as means ± SEM.
DNA fragmentation detection assay
Cell pellets (treated with 0 or 35 µM of emodin after 72 h) were lysed with 600 µL nuclear lysis buffer (10 mM Tris-HCl pH 7.5, 400 mM NaCl, 100 mM EDTA, 0.6% SDS) and 10 µL RNase (4 mg/mL) and incubated in water bath at 37 °C for 5 min. After gentle mixing, 200 µL of protein precipitation solution (6 M NaCl) was added and chilled on ice for 5 min. Next, the mixture was centrifuged at 17 779 g for 10 min (at room temperature), and the supernatant was collected. The supernatant was subsequently mixed with 600 µL isopropanol and chilled on ice for 15 min. Next, this mixture was centrifuged at 17 779 g for 20 min, and the pellet was washed with 600 µL of 70% ethanol. The DNA pellet was air dried and re-suspended in 200 µL of Tris-EDTA buffer [10 mM Tris-HCl (pH 8.0) and 1 mM EDTA], and the concentration of DNA was determined spectroscopically. DNA (10 µg/µL) was electrophoresed on 1.5% agarose gel with ethidium bromide at 100 V for 2 h and analyzed. The fragmentized inter-nucleosomal DNA was visualized using a UV transilluminator.
Assessment of mRNA expression levels in apoptosis-related genes by real-time PCR
To determine the gene expression levels of Fas ligand (FASL), myeloid cell leukemia sequence 1 (MCL1), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), BCL2-associated X protein (BAX), cyclin D1 (CCND1) and v-myc myelocytomatosis viral oncogene homolog (C-MYC), untreated or emodin (35 µM)-treated MCF-7 cells were trypsinized, and the total RNA was extracted by TRIzol reagent (Molecular Research Center, Inc., Cincinnati, OH). Next, the RNA was reverse transcribed into cDNA by real-time (RT-PCR) (MBI Fermentas) using the SuperScript First-Strand synthesis system (Invitrogen, Life Technologies Inc., Gaithersburg, MD). RT-PCR was performed with a SYBR Green real-time detection kit (Roche Diagnostics, Sydney, Australia) using the Cepheid SmartCycler (Sunnyvale, CA). Total reaction mixture (25 µL) containing cDNA, iQ SYBR Green Supermix, 10 µm forward primer and reverse primer (), and sterile milli-Q water was prepared. The SYBR Green Supermix (Invitrogen) contained dNTPs (0.4 mM of each), Taq polymerase (50 units/mL), MgCl2 (6 mM), KCl (100 mM) and Tris-HCl at pH 8.4 (40 mM). Expression levels of cDNA were compared to the internal standard ribosomal protein S9 (RPS9), a housekeeping gene, to correct for differences in the RNA quantity of the samples used. RT-PCR reaction procedures were as follows: 50 °C for 2 min (UDG incubation), 95 °C for 3 min (UDG inactivation and DNA polymerase activation), 40 PCR cycles (except FASL with 50 PCR cycles) consisting of denaturation for 10 s at 95 °C, annealing and extension for 30 s (at 60 °C for RPS9, GAPDH, BAX, CCND1; at 62 °C for MCL1 at 65.7 °C for FASL and 67 °C for C-MYC) and primer extension at 72 °C for 30 s. The internal melting curves of the final PCR products were analyzed from 60 °C to 95 °C. The mRNA expression level of interest was related to the internal standard, and RPS9 was used to correct the differences between the quantity and quality of different samples of RNA. The threshold cycle for each amplification cycle was found and plotted against the standard curve to calculate the concentration of gene product. Values from each group were normalized to the RPS9 gene product. A standard curve assessing the level of DNA was generated for each gene. Each RT-PCR reaction was performed in triplicate.
Table 1. Names and accession number of genes. Forward and reverse primer sequence and product size of genes.
Statistical analysis
All data are presented as means ± standard errors of mean (SEM). Statistical analysis was performed by analysis of variance (ANOVA) to detect significant differences in multiple comparisons with the Bonferroni post hoc test and comparing all pairs of columns. A value of probability (p) < 0.05 was considered to be statistically significant. All statistical analyses were performed using GraphPad Prism 4.02 software for Windows (GraphPad Software, San Diego, CA).
Results
Inhibitory effect on cell growth
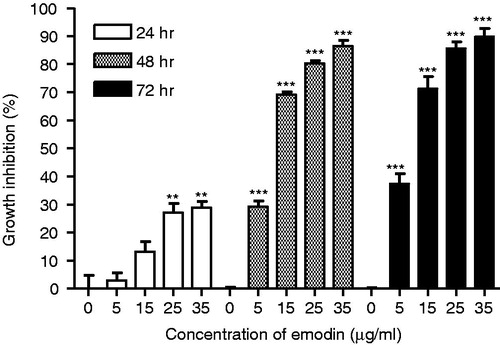
To test the growth inhibitory effects of emodin on MCF-7 cells and to estimate the IC50 values, additional concentration points (0, 5, 10, 15, 20, 25, 30 and 35 µg/mL) were used. MCF-7 cells treated with emodin showed significant growth inhibitory effects after 72 h of treatment with the IC50 value equal to 7.22 µg/mL (∼30 µM; ). To investigate whether the cytotoxic effect of emodin is time- and concentration-dependent, MCF-7 cells were treated with different concentrations (0, 5, 15 and 35 µg/mL) of emodin for 24, 48 and 72 h. The data indicate that the growth inhibitory effects of emodin on MCF-7 cells are both time- and concentration-dependent ().
Figure 1. Growth inhibitory effect of emodin on MCF-7 cells after the 72-h treatment. Data are expressed as means ± SEM, n = 3.
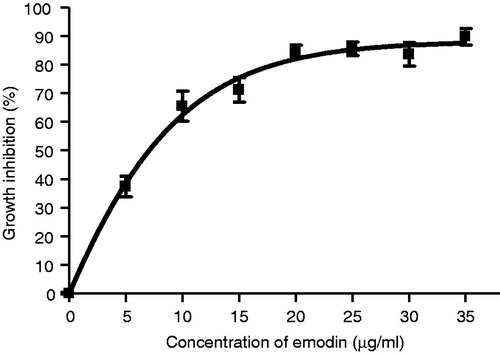
Figure 2. Growth inhibitory effect of emodin (0, 5, 15, 25 and 35 µg/mL) on MCF-7 cells after 24, 48 and 72 h treatments. Data are expressed as means ± SEM, n = 3. **p < 0.01 and ***p < 0.001 represent significant differences when compared with their corresponding control group (emodin, 0 µg/mL).
Inhibition on colony formation
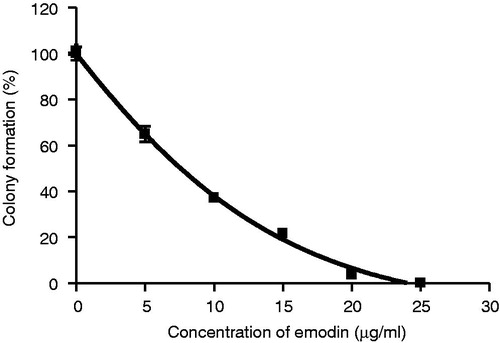
A narrower concentration range with a finer scale (0, 5, 10, 15, 20 and 25 µg/mL) was subsequently used to determine differences in the sensitivity of the colony formation assay. As shown in , emodin treatment was found to exert a concentration-dependant inhibitory effect on the colony-forming ability of MCF-7 cells with IC50 values equal to 7.60 µg/mL (∼30 µM).
Figure 3. Inhibitory effect of emodin on the formation of colony of MCF-7 cells after the 10-d treatment. Data are expressed as means ± SEM, n = 3.
Induction of single-strand DNA damage
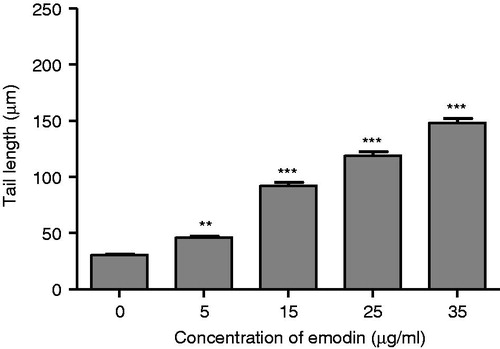
The COMET assay was performed to study DNA damage in the emodin-treated MCF-7 cells after undergoing the 72-h treatment. The COMET assay is a sensitive method for monitoring single strand (ss) DNA breaks at the single-cell level. Any DNA damage is represented as an increase in the tail length (tail migration) of the DNA strand. When MCF-7 cells were treated with 0, 5, 15, 25 or 35 µg/mL of emodin for 72 h, ssDNA damage was significant, as indicated by the increased tail length observed in the emodin-treated cells compared with the controls (). Moreover, the DNA damage induced by the emodin treatment was dose-dependent.
Figure 4. Effect of emodin (0, 5, 15, 25 and 35 µg/mL) on DNA damage in MCF-7 cells after the 72-h treatment represented as comet tail length (µm). Data are expressed as mean ± SEM, n = 100. **p < 0.01 and ***p < 0.001 represent significant differences when compared with the control group.
Induction of chromosomal DNA fragmentation
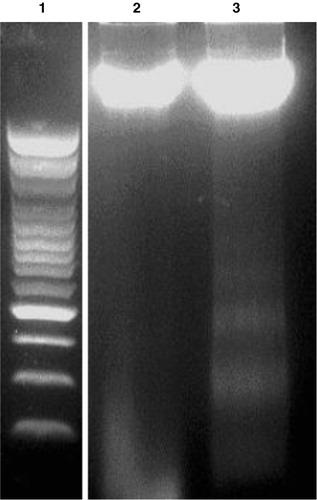
The IC50 value (30 µM) obtained from a cell inhibition assay was used to validate the emodin-induced apoptotic responses in MCF-7 cells. Chromosomal DNA from untreated and emodin-treated (30 µM) MCF-7 cells was isolated, extracted, and resolved using agarose gel electrophoresis. The gel was analyzed under a UV transilluminator to detect the presence of any fragmented DNA. Emodin treatment resulted in inter-nucleosomal DNA cleavage in MCF-7 cells as indicated by DNA laddering, while the inter-nucleosomal DNA from untreated cancer cells remained intact ().
Figure 5. Chromosomal DNA of emodin-treated or -untreated MCF-7 cells following electrophoresis through a 1.5% agarose gel under UV illuminator. Lane 1: 100 bp DNA Marker; lane 2: DNA from 0.05% DMSO-treated MCF-7 cells; lane 3: DNA from 0.05% DMSO and 30 µM emodin-treated MCF-7 cells. All MCF-7 cells were treated with emodin for 72 h.
Modulation of mRNA levels of apoptosis-related genes
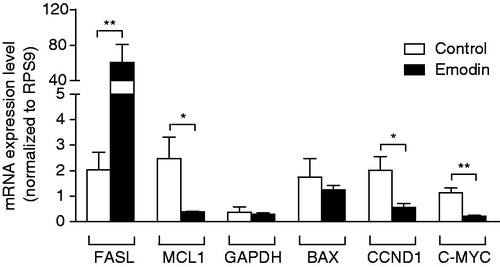
To further investigate the apoptotic effects of emodin on MCF-7 cells after emodin (30 µM, the IC50 value) treatment for 72 h, the mRNA levels of apoptosis-related genes, namely FASL, MCL1, GAPDH, BAX, CCND1 and C-MYC, were analyzed. By comparing the mRNA levels of the genes between the control and the emodin-treated samples, the effect of emodin on the gene expression of the apoptosis-related genes could be examined. All the mRNA levels of genes were normalized to that of the internal standard RPS9.
As shown in , the 72-h emodin treatment could significantly up-regulate the gene expression of FASL (p < 0.01) and significantly down-regulate the expression of MCL1 (p < 0.05), CCND1 (p < 0.05) and C-MYC (p < 0.01) as compared with those of the control. However, there were no significant changes in the gene expression of GAPDH and BAX (both p > 0.05) in MCF-7 cells after the 72-h emodin treatment.
Figure 6. RT-PCR analysis of the gene expression of FASL, MCL1, GAPDH, BAX, CCND1 and C-MYC on MCF-7 cells after the 72-h treatment with emodin (0 or 30 µg/mL). The expression level of each gene was normalized to that of the RPS9 gene. Data are expressed as means ± SEM, n = 3. *p < 0.05 and **p < 0.01 represent significant differences when compared with the control.
Discussion
In this study, the in vitro anticancer effect of emodin on the human breast carcinoma MCF-7 cells was investigated by monitoring the cell growth, colony formation, changes of chromosomal DNA, and mRNA levels of apoptosis-related genes in emodin-treated cancer cells. This study confirmed that emodin has strong dose- and time-dependent anticancer activities against MCF-7 cells () with an IC50 equal to 7.22 µg/mL (∼30 µM; ). This finding suggests that emodin functions by inhibiting the growth of tumor cells. In fact, inhibition of the growth of tumor cells is one of the major benchmark of successful cancer treatment. Emodin also inhibits the colony growth potential of MCF-7 cells in a dose-dependent manner (). This result suggests that emodin could inhibit the anchorage-independent growth of MCF-7 cells.
Apoptosis and necrosis are two types of cell death. In this study, the emodin-treated MCF-7 cells showed cell shrinkage (data not shown), DNA single-strand breakage at the single-cell level (), and chromosomal DNA fragmentation (), which are morphological hallmarks of apoptosis. These findings suggest that emodin could activate the activity of the DNA endonuclease on double-strand cleavage of chromosomal DNA into fragments. Similar observations have been reported in emodin-treated human lung squamous cell carcinoma CH2 (Lee, Citation2001; Lee et al., Citation2001), the human hepatoma cell lines HepG2 and C3A (Shieh et al., Citation2004), human promyeloleukemic HL-60 cells (Chen et al., Citation2002), human cervical cancer Bu 25TK cells (Srinivas et al., Citation2003), and human cancer HSC5 and MDA-MB-231 cells (Huang et al., Citation2004).
In fact, apoptosis can be triggered by a variety of physiological and stress stimuli that initiate changes in gene regulation through one or more distinct signaling pathways (Ishitani et al., Citation2003; Zhan et al., Citation1994). These stimuli may modulate the gene expression that activates various signaling pathways in apoptosis (Cameron & Feuer, Citation2001). In this study, the mRNA expression levels of genes that are involved in apoptosis, namely, FASL, MCL1, GAPDH, BAX, CCND1 and C-MYC, were investigated. All gene expressions were normalized to the reference gene RPS9.
The current study demonstrated that the mRNA level of FASL was significantly elevated in MCF-7 cells () after treatment of emodin for 72 h. FASL is the gene that encodes the Fas ligand (CD95 ligand). Fas ligand is a membrane glycoprotein that belongs to the tumor necrosis factor family (TNF family) and is involved in the extrinsic pathways of apoptosis. The up-regulation of FASL gene expression enhanced apoptosis through the extrinsic pathways which in turn causes internucleosomal DNA fragmentation and other structural alternations (Cameron & Feuer, Citation2001). The up-regulation of FASL in emodin-treated cells implies that emodin could inhibit the growth of MCF-7 cells through the Fas-mediated apoptotic pathway. Similar observations have been reported in emodin-treated human hepatoma cells (Shieh et al., Citation2004).
Mcl-1 and Bax belong to the Bcl-2 family of proteins. Mcl-1 refers to myeloid cell leukemia sequence 1 that plays a key role in the regulation of cell death with particular members, such as Bcl-2, and serves as an anti-apoptotic activator that inhibits apoptosis through blocking cytochrome c release from the mitochondria in an intrinsic pathway. The down-regulation of MCL1 gene expression leads to apoptosis. The BAX gene encodes the Bcl-2-associated X protein that forms a heterodimer with Bcl-2. Bax functions as an apoptosis activator and regulates apoptosis in the intrinsic pathway. Up-regulation of BAX gene expression results in apoptosis. In the present study, emodin induced the down-regulation of MCL1 expression but did not change the gene expression of BAX in MCF-7 cells after treatment for 72 h (). These data suggested that the apoptotic effect of emodin is through the down-regulation of MCL1 expression but not the up-regulation of BAX expression. In the MCL1 pathway, cytochrome c released from the mitochondria in an intrinsic pathway was promoted. This activity results in the formation of a complex in the cytoplasm that activates caspase-9 and forms an apoptosome. The apoptosome then activates caspase-3, which in turn activates the caspase cascade and results in degradation (Burlacu, Citation2003; Gross et al., Citation1999; Link & Harrison, Citation2001). In the literature, there is no uniform effect of emodin treatment on BAX expression. Some studies showed that there was an up-regulation in BAX expression in emodin-treated cells (Lee, Citation2001; Lee et al., Citation2001; Su et al., Citation2005); while others reported that BAX gene expression remained unchanged in emodin-treated human promyeloleukemic HL-60 cells (Chen et al., Citation2002). Further studies are warranted to explain the discrepancy.
GAPDH is a gene that encodes glyceraldehyde-3-phosphate dehydrogenase, a key enzyme, and catalyzes an important energy-yielding step in glycolysis, the reversible oxidative phosphorylation of glyceraldehyde-3-phosphate in the presence of inorganic phosphate and nicotinamide adenine dinucleotide. GAPDH is an abundant RNA species; therefore, it is often used as a potential internal RNA standard or housekeeping gene. However, it has been reported that a wide variation in GAPDH expression was observed in different tissues, such as insulin-, dexamethasone- and mitogen-treated cells, virally transformed or oncogene-transfected fibroblasts, and human pancreatic or colon adenocarcinomas (Mogal & Abdulkadir, Citation2006). In addition, recent advances suggest that GAPDH exhibits diverse non-glycolytic functions depending on its subcellular localization. One of the most intriguing possible functions is the induction of apoptosis. Over-expression of GAPDH was reported to induce apoptosis through the initiation of one or more apoptotic cascades (Ishitani et al., Citation2003). In the current study, however, emodin treatment brought no significant changes in GAPDH expression in MCF-7 cells (), suggesting that emodin-induced apoptosis is GAPDH-independent.
This report is the first study to demonstrate that emodin could lower the expression of CCND1 and C-MYC in MCF-7 cells after treatment for 72 h (). Down-regulation of CCND1 and C-MYC blocks the transition of cells from the G1 phase to the S phase, thereby causing cells to arrest in the G0/G1 phase of the cell cycle (Liu et al., Citation2005; Pelengaris et al., Citation2002a,Citationb). This G0/G1 phase arrest in the cell cycle is an irreversible process in which the cells are unable to repair damages and ultimately undergo apoptosis. This finding explains how emodin inhibits growth and induces apoptosis in MCF-7 cells.
In summary, emodin could induce growth inhibition and apoptosis in MCF-7 cells through the modulation of expression of apoptosis-related genes. Emodin treatment up-regulated FASL gene expression but down-regulated the expression of MCL1, CCND1 and C-MYC. The growth inhibitory effects of emodin might involve both the intrinsic and the extrinsic apoptotic pathways and cell cycle arrest. However, further experiments are needed to elucidate the mechanisms of the activities of emodin.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Acknowledgements
We thank the Shenzhen Municipal Key Laboratory Advancement Program, Shenzhen, People’s Republic of China for its support to the project and Ms. Josephine Hong Man Leung for proofreading the manuscript.
Related Research Data
References
- Ahn KS, Hahn BS, Kwack K, et al. (2006). Platycodin D-induced apoptosis through nuclear factor-kappaB activation in immortalized keratinocytes. Eur J Pharmacol 537:1–11
- Basu S, Ghosh A, Hazra B. (2005). Evaluation of the antibacterial activity of Ventilago madraspatana Gaertn., Rubia cordifolia Linn. and Lantana camara Linn.: Isolation of emodin and physcion as active antibacterial agents. Phytother Res 19:888–94
- Burlacu A. (2003). Regulation of apoptosis by Bcl-2 family proteins. J Cell Mol Med 7:249–57
- Cameron R, Feuer G. (2001). The effect of drugs and toxins on the process of apoptosis. Drug Metabol Drug Interact 18:1–32
- Chang GC, Hsu SL, Tsai JR, et al. (2004). Molecular mechanisms of ZD1839-induced G1-cell cycle arrest and apoptosis in human lung adenocarcinoma A549 cells. Biochem Pharmacol 68:1453–64
- Chen YC, Shen SC, Lee WR, et al. (2002). Emodin induces apoptosis in human promyeloleukemic HL-60 cells accompanied by activation of caspase 3 cascade but independent of reactive oxygen species production. Biochem Pharmacol 64:1713–24
- Gross A, McDonnell JM, Korsmeyer SJ. (1999). BCL-2 family members and the mitochondria in apoptosis. Genes Dev 13:1899–911
- Huang HC, Chu SH, Chao PD. (1991a). Vasorelaxants from Chinese herbs, emodin and scoparone, possess immunosuppressive properties. Eur J Pharmacol 198:211–13
- Huang HC, Lee CR, Chao PD, et al. (1991b). Vasorelaxant effect of emodin, an anthraquinone from a Chinese herb. Eur J Pharmacol 205:289–94
- Huang Q, Shen HM, Ong CN. (2004). Inhibitory effect of emodin on tumor invasion through suppression of activator protein-1 and nuclear factor-kappaB. Biochem Pharmacol 68:361–71
- Huang Z, Chen G, Shi P. (2008). Emodin-induced apoptosis in human breast cancer BCap-37 cells through the mitochondrial signaling pathway. Arch Pharm Res 31:742–8
- Ishitani R, Tajima H, Takata H, et al. (2003). Proapoptotic protein glyceraldehyde-3-phosphate dehydrogenase: A possible site of action of antiapoptotic drugs. Prog Neuropsychopharmacol Biol Psychiatry 27:291–301
- Jang BC, Paik JH, Jeong HY, et al. (2004). Leptomycin B-induced apoptosis is mediated through caspase activation and down-regulation of Mcl-1 and XIAP expression, but not through the generation of ROS in U937 leukemia cells. Biochem Pharmacol 68:263–74
- Kim MS, Park MJ, Kim SJ, et al. (2005). Emodin suppresses hyaluronic acid-induced MMP-9 secretion and invasion of glioma cells. Int J Oncol 27:839–46
- Klaude M, Eriksson S, Nygren J, Ahnstrom G. (1996). The comet assay: Mechanisms and technical considerations. Mutat Res 363:89–96
- Kummalue T, Oc P, Jiratchariyakul W, et al. (2007). Antiproliferative effect of Erycibe elliptilimba on human breast cancer cell lines. J Ethnopharmacol 110:439–43
- Lee EJ, Min HY, Chung HJ, et al. (2005). A novel adenosine analog, thio-Cl-IB-MECA, induces G0/G1 cell cycle arrest and apoptosis in human promyelocytic leukemia HL-60 cells. Biochem Pharmacol 70:918–24
- Lee HZ. (2001). Effects and mechanisms of emodin on cell death in human lung squamous cell carcinoma. Br J Pharmacol 134:11–20
- Lee HZ, Hsu SL, Liu MC, Wu CH. (2001). Effects and mechanisms of aloe-emodin on cell death in human lung squamous cell carcinoma. Eur J Pharmacol 431:287–95
- Li HL, Chen HL, Li H, et al. (2005). Regulatory effects of emodin on NF-kappaB activation and inflammatory cytokine expression in RAW 264.7 macrophages. Int J Mol Med 16:41–7
- Li WY, Chan SW, Guo DJ, et al. (2009). Water extract of Rheum officinale Baill. induces apoptosis in human lung adenocarcinoma A549 and human breast cancer MCF-7 cell lines. J Ethnopharmacol 124:251–6
- Lin HI, Lee YJ, Chen BF, et al. (2005). Involvement of Bcl-2 family, cytochrome c and caspase 3 in induction of apoptosis by beauvericin in human non-small cell lung cancer cells. Cancer Lett 230:248–59
- Link M, Harrison DJ. (2001). To live or die – A cell's choice. Essays Biochem 37:109–20
- Liu T, Zhu E, Wang L, et al. (2005). Abnormal expression of Rb pathway-related proteins in salivary gland acinic cell carcinoma. Hum Pathol 36:962–70
- Mogal A, Abdulkadir SA. (2006). Effects of histone deacetylase inhibitor (HDACi); trichostatin-A (TSA) on the expression of housekeeping genes. Mol Cell Probes 20:81–6
- Pelengaris S, Khan M, Evan GI. (2002a). c-MYC: More than just a matter of life and death. Nat Rev Cancer 2:764–76
- Pelengaris S, Khan M, Evan GI. (2002b). Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 109:321–34
- Petty WJ, Dragnev KH, Dmitrovsky E. (2003). Cyclin D1 as a target for chemoprevention. Lung Cancer 41:S155–61
- Shieh DE, Chen YY, Yen MH, et al. (2004). Emodin-induced apoptosis through p53-dependent pathway in human hepatoma cells. Life Sci 74:2279–90
- Singh NP, McCoy MT, Tice RR, Schneider EL. (1988). A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res 175:184–91
- Srinivas G, Anto RJ, Srinivas P, et al. (2003). Emodin induces apoptosis of human cervical cancer cells through poly(ADP-ribose) polymerase cleavage and activation of caspase-9. Eur J Pharmacol 473:117–25
- Su YT, Chang HL, Shyue SK, Hsu SL. (2005). Emodin induces apoptosis in human lung adenocarcinoma cells through a reactive oxygen species-dependent mitochondrial signaling pathway. Biochem Pharmacol 70:229–41
- Way TD, Kao MC, Lin JK. (2005). Degradation of HER2/neu by apigenin induces apoptosis through cytochrome c release and caspase-3 activation in HER2/neu-overexpressing breast cancer cells. FEBS Lett 579:145–52
- Wu SJ, Ng LT, Lin CC. (2005). Cinnamaldehyde-induced apoptosis in human PLC/PRF/5 cells through activation of the proapoptotic Bcl-2 family proteins and MAPK pathway. Life Sci 77:938–51
- Zhan Q, Fan S, Bae I, et al. (1994). Induction of Bax by genotoxic stress in human cells correlates with normal p53 status and apoptosis. Oncogene 9:3743–51
- Zhang CX, Huang KX. (2006). Mechanism of apoptosis induced by a polysaccharide, from the loach Misgurnus anguillicaudatus (MAP) in human hepatocellular carcinoma cells. Toxicol Appl Pharmacol 210:236–45
- Zhang HQ, Zhou CH, Wu YQ. (2005). Effect of emodin on small intestinal peristalsis of mice and relevant mechanism. World J Gastroenterol 11:3147–50