
Abstract
Background: Carbamazepine, which was developed primarily for the treatment of epilepsy, is now also useful for the treatment of non-epileptic disorders and inflammatory hyperalgesia. However, the mechanism of its anti-neuroinflammatory action remains poorly understood.
Objective: This study elucidates the anti-neuroinflammatory capacity of carbamazepine on microglial activation and the relative mechanisms involved.
Materials and methods: The microglial BV-2 cells were pretreated with carbamazepine for 15 min before activation by lipopolysaccharide (LPS). After LPS stimulation, the expression of inducible nitric oxide synthase (iNOS) was analyzed by Western blotting (WB) and reverse transcription-polymerase chain reaction. Signaling proteins and cyclooxygenase (COX)-2 were also evaluated by WB. The levels of nitrate and tumor necrosis factor (TNF)-α were analyzed by the Griess method and enzyme-linked immunosorbant assay, respectively. The formation of intracellular reactive oxygen species (ROS) was examined by fluorescent analysis.
Results: Carbamazepine strongly attenuated LPS-induced production of NO and iNOS protein at concentrations of 5, 10, and 20 μM. Consistently, it could markedly suppress iNOS mRNA expression stimulated by LPS. Among the signaling pathways, LPS-mediated IκBα degradation or JNK MAPK phosphorylation was not affected by carbamazepine. Interestingly, it was found that carbamazepine could concentration-dependently inhibit LPS-activated phospho-Akt expression. Nevertheless, LPS-induced ROS production was not affected by carbamazepine. Carbamazepine (20 μM) affected either COX-2 expression or TNF-α production induced by LPS with approximately 70% and 51% inhibition, respectively.
Discussion and conclusion: Our findings showed that carbamazepine exerted selective inhibition on LPS-induced microglial iNOS expression through the down-regulation of Akt activation, and thus may play a pivotal role of anti-neuroinflammation in its therapeutic efficacy.
Introduction
Neuroinflammation is suggested to be an important pathological factor in brain diseases, and may contribute to epileptogenesis (Vezzani & Granata, Citation2005). Pathological insults to the central nervous system induce a neuroinflammatory response characterized by marked activation of astrocytes and microglia, acute up-regulation of pro-inflammatory mediators, and finally damage to the neurons and disrupt blood–brain barrier in a variety of neuropathological disorders, such as brain trauma, epilepsy and stroke (Choi et al., Citation2009; Lloyd et al., Citation2008; Wang, Citation2010). Among these proinflammatory mediators, extensive NO is converted from arginine by the catalyzing action of iNOS. The iNOS has been implicated in the neurodegenerative processes (Brown & Bal-Price, Citation2003), since it produces large amounts of NO continuously for long periods, a feature that is responsible for the damage in the nervous and peri-cerebral vascular systems.
NO is involved in a number of pathological conditions of the brain, such as multiple sclerosis, cerebral ischemia, and epileptic seizure. NO activation was significantly found in the hippocampus and other brain regions following different chemical-induced status epilepticus (Kim et al., Citation2010; Liu et al., 2010a). It is well known that NO plays an important role in the pro-inflammatory responses, and the overwhelming release of NO may trigger a series of inflammatory cascades in the CNS from activated microglial cells (Stoll & Jander, Citation1999). Multiple signaling effectors are activated in response to endotoxic lipopolysaccharide (LPS) in the microglial cells (Lehnardt et al., Citation2003). Therefore, LPS has been widely used as an inflammatory agent to create neuro-inflammation through its stimulation of iNOS and cyclooxygenase-2 expression in the brain (Oh et al., Citation2009). It was demonstrated that the signaling pathways involved in the induction of microglial iNOS and NO production are partly identified to be due to the activation of NF-κB (Jin et al., Citation2006), phosphatidylinositol 3-kinase (PI3K)-Akt (Kim et al., Citation2004; Liu et al., 2010b), mitogen-activated protein kinases (MAPKs) (Bhat & Fan, Citation2002), and reactive oxygen species (ROS) (Pawate et al., Citation2004).
In contrast, the efficacy of some anticonvulsants could be potentiated by pharmacological NOS inhibition (De Sarro et al., Citation2000). Consistently, iNOS knockout mice could attenuate the kindled status induced by pentylenetetrazole when compared with the wild-type mice (De Luca et al., Citation2006). Recently, it has been demonstrated that methylmalonate-induced seizures been attenuated in iNOS knockout mice (Ribeiro et al., Citation2009). Therefore, blockade of these pro-inflammatory pathways followed by the down-regulation of NO and iNOS levels in microglial cells may be an attractive therapeutic strategy against neurodegenerative diseases and epileptics.
Carbamazepine is widely used for the treatment of epilepsy and neuropsychiatric disorders (Trimble, Citation2002). Reduction of firing rate in voltage-gated sodium channels is the major function of carbamazepine (Macdonald & Kelly, Citation1995). Although reported infrequently, some adverse drug reactions have been remarked during the treatment of carbamazepine (Phillips & Mallal, Citation2009). Therefore, the indications and therapeutic effects of this psychiatric drug are not clearly elucidated. A previous study has demonstrated that the suppressive effect of carbamazepine on adenylyl cyclase 5 within dopaminergic brain regions is associated with the antidepressant effect (Mann et al., Citation2009). Carbamazepine also exerted a protective effect on the neuronal hyperplasia and abnormal gene expression in megalencephaly (Almgren et al., Citation2008). Additionally, it was found that the expression of renal aquaporin 2 (De Bragança et al., Citation2010) and retinoic acid-induced functional sodium iodide symporter (Willhauck et al., Citation2010) were induced by carbamazepine. Interestingly, this drug exerted anti-proliferative action on Vero cells (Pérez Martín et al., Citation2008), but increased CD4+ T cell level in HIV/AIDS patients (Lee et al., Citation2010). Furthermore, epithelial calcium transport (Von Borstel Smith et al., Citation2007) and acetylcholinesterase (Siebel et al., Citation2010) were attenuated by carbamazepine. Some reports also indicated that it could abrogate inflammatory hyperalgesia and non-epilepsy disorders (Iwamoto et al., Citation2011; Johannessen, Citation2008), but its anti-inflammatory mechanisms remained poorly understood. In contrast, the keto-derivative of carbamazepine, oxcarbazepine, exerted the inhibitory effects on the production of interleukin (IL)-1β, IL-2, and tumor necrosis factor-α from toxin-activated human whole blood (Himmerich et al., Citation2013). This derivative also been reported to slow the accumulation of disability in experimental autoimmune encephalomyelitis (Al-lzki et al., Citation2014) and attenuate inflammatory hyperalgesia (Stepanovic-Petrovic et al., Citation2011).
Our hypothesis is that carbamazepine counteracts neuroinflammation via influence on iNOS expression in the microglial cells. In this study, we evaluated the effects of carbamazepine on microglial functions and its inhibitory mechanism on iNOS expression in activated microglial cells.
Materials and methods
Reagents
Carbamazepine, LPS, thiazolyl blue tetrazolium bromide (MTT), 4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid (HEPES), sodium dodecylsulfate (SDS), leupeptin, aprotinin, sodium fluoride, sodium orthovanadate, sodium pyrophosphate, phenylmethylsulfonyl fluoride (PMSF), diethyl pyrocarbonate (DEPC), bovine serum albumin (BSA), 2′,7′-dichlorofluorescin diacetate (DCFH-DA), apocynin, sodium nitrate, and phorbol 12-myristate 13-acetate (PMA) were all obtained from Sigma-Aldrich (St. Louis, MO). Anti-mouse and anti-rabbit immunoglobulin G (IgG)-conjugated horseradish peroxidase (HRP) was purchased from Amersham Biosciences (Sunnyvale, CA) and/or Jackson-ImmunoResearch (West Grove, PA). The rabbit polyclonal antibody (pAb) specific for either mouse iNOS or IκBα were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). A pAb specific for mouse COX-2 was purchased from Cayman (Ann Arbor, MI). The anti-phospho-c-Jun N-terminal kinase (JNK) pAb and the anti-Akt pAb were obtained from Cell Signaling (Beverly, MA). The Hybond-P PVDF membrane, ECL Western blotting detection reagent, and analysis system were purchased from Amersham (Buckinghamshire, UK). Carbamazepine was dissolved in dimethyl sulfoxide (DMSO, ≥ 99.9%) with a constant final concentration of 0.2% (v/v). All other chemicals were used as the reagent grade.
Cell cultivation
The mouse BV-2 microglia cell line was a kind gift from Professor Wang-Wang Lin (National Taiwan University, Taiwan). Cells were cultured in DMEM supplemented with l-glutamine (3.65 mM), penicillin (90 units/ml), streptomycin (90 μg/ml), HEPES (18 mM), NaHCO3 (23.57 mM), and 10% heat-inactivated fetal bovine serum (FBS) at 37 °C in humidified air with 5% CO2. For stimulation of LPS, BV-2 cells were seeded at 2.5 × 106 per well of Costar 6-well tissue culture plates in complete media until a confluence of 85% was reached. After 24 h, cells were changed to serum (0.5%) media. Cells were treated with vehicle or indicated concentrations of carbamazepine (1, 5, 10, and 20 μM) for 15 min, and then cells were treated with LPS for another 24 h. At the end of incubation period, the remaining cells were disrupted by the lysing buffer (50 mM HEPES, 50 mM NaCl, 5 mM EDTA, 10 μg/ml aprotinin, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 2 mM dithiothreitol, 5 mM sodium pyrophosphate, 1 mM Na3VO4, 10 mM NaF, 1% Triton X-100, pH 7.4). Lysates were centrifuged at 13 400 × g at 4 °C for 20 min and their supernatants were finally collected and stored at −80 °C for immunoblotting assay as described previously (Chou et al., Citation2010).
Western blot analysis
Immunoblotting analyses were performed as previously described (Chou et al., Citation2010). Cellular supernatants from each sample were quantified by the Bio-Rad Protein Assay Dye®, using bovine serum albumin as a standard. Thereafter, samples were mixed with 6 × sample buffer (0.35 M Tris, 10% w/v SDS, 30% v/v glycerol, 0.6 M DTT, and 0.012% w/v bromophenol blue, pH 6.8) and heated to 95 °C for 5 min. The proteins were separated by electrophoresis and transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% non-fat milk in TBS-0.1% Tween 20 and sequentially incubated with primary antibodies and HRP-conjugated secondary antibodies, followed by enhanced chemiluminescence (ECL) detection (Amersham Biosciences, Sunnyvale, CA). The Digital Scanning System with BIO-PROFIL Bio-1D light analytical software (V2000, Vilber Lourmat, Marue La Vallee, France) was used for the quantitative densitometric analysis. Data of specific protein levels are presented as relative folds in relation to the control.
Determination of nitrate levels
The concentrations of nitrate in the conditioned medium of BV-2 cells were measured by using the Nitrate/nitrite Colorimetric Assay Kit® (Cayman, Ann Arbor, MI). The assay was performed according to the manufacturer’s instructions. The concentration of nitrate in each sample was calculated by a standard curve of sodium nitrate.
Cellular viability assay
The cytotoxic effect of carbamazepine (1, 5, 10, and 20 μM) against the BV-2 cell line was determined by the MTT method as described previously (Chung et al., Citation2009). The passage (proliferation cycle) of the cells with 0.5% FBS were not in the exponential growth phase. Briefly, after incubation with test reagents for 22 h, the MTT solution was added to each well as a final concentration of 0.5 mg/ml. Additional 2 h later, the supernatant was discarded and replaced with DMSO, the dissolved formazan product was measured at 550 nm by an ELISA reader (MRX Microplate Reader, Molecular Devices, Sunnyvale, CA). The following formula was used to calculate the cell viability (%, absorbance of the experiment samples/absorbance of the control × 100%).
Reverse transcription-PCR assay
Reverse transcription-PCR was performed on total RNA from BV-2 cells, as previously described by Hsiao et al. (Citation2004) with some modification. Total RNA was extracted from BV-2 cells by TRIzol (Invitrogen, Carlsbad, CA) and quantified spectrophotometrically (GeneQuant pro UV/Vis spectrophotometer, Amersham). An aliquot of total RNA was reverse transcribed and amplified by PCR (Super Script One-Step RT-PCR system kit, Gibco, Invitrogen, Carlsbad, CA). Briefly, the reaction mixture contained 0.5 μg of the RNA sample and 0.2 μM of each primer, in a total volume of 50 μl and indicated cycles (GeneAmp PCR 2400, Perkin-Elmer, Carlsbad, CA). The reverse transcription was performed at 50 °C for 30 min and 94 °C for 2 min. For iNOS system, each cycle consisted of 20 s at 95 °C. Therefore, 30 s at 57 °C and finally 30 s at 72 °C for total 23 cycles. For β-actin system, each cycle consisted of 20 s at 95 °C. Therefore, 30 s at 47 °C and finally 20 s at 72 °C for total 30 cycles. The final extension condition consisted of 5 min at 72 °C, and thereafter 4 °C. Specific primers used for iNOS were forward (F): 5′-GTGCTGAAGGACACACTAAAGAAGA-3′ and reverse (R): 5′-TTGCCATCCTCTCAAAGTTGTAGG-3′; and for β-actin were F: 5′-TGGAATCCTGTGGCATCCATGGAAAC-3′ and R: 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′. Each amplified cDNA was assessed by agarose gel electrophoresis. The quantitative densitometric analyses on gel bands were performed by the Photo-Print Digital Imaging System (IP-008-SP) with analytical software (BIO-PROFIL Bio-1D light, V2000). The indicated bands were quantified according to their folds of relative intensities.
Assay of intracellular ROS
BV-2 cells were grown in the phenol-red-free medium with FBS (10%) in 96-well culture plate (2.5 × 105 cells/ml, 200 μl/well). After 24 h, the medium was changed into Hank’s Balanced Salt Solution (HBSS) with 0.25% BSA and labeled with DCFH-DA (20 μM) for another 1 h incubation. Thereafter, fresh HBSS was changed into the indicated well with vehicle or test compounds (carbamazepine or apocynin, 20 μM). BV-2 cells were stimulated with LPS (150 ng/ml) or PMA (10 nM) for 2 h at 37 °C. After stimulation, the labeled cells were carefully measured by 96-well fluorescent microplate and read at excitation/emission wavelengths of 485/530 nm in a microplate multimode reader (Varioskan Flash®, Theremo Scientific, Waltham, MA). The data of ROS levels were presented as relative folds to fluorescent intensity of control.
Analysis of TNF-α by ELISA
BV-2 cells were treated with LPS in the presence or absence of carbamazepine (5, 10, and 20 μM) for 48 h, and the culture medium from each experiment was collected. The concentration of TNF-α released by BV-2 cells into the medium was measured using the mouse TNF-α ELISA kit® (e-Bioscience, Amersham, UK), according to the manufacturer’s instruction.
Statistical analysis
Data are presented as the means ± S.E.M with the number of experiments indicated. For the analysis of the results, a one-way analysis of variance (ANOVA) test was performed using the Sigma Stat v3.5 software (Systat Software, Chicago, IL). The Student–Newman–Keuls test was subsequently used as a p value less than 0.05 being considered statistically significant.
Results
The effects of carbamazepine on NO release and iNOS production
We first studied iNOS production induced by various concentrations of LPS in microglial (BV-2) cells prior to the evaluation of the effects of carbamazepine. After stimulation for 24 h, intracellular iNOS was detected by immunoblotting analysis (). According to these results, 150 ng/ml of LPS was selected as a suitable concentration for further experiments. After stimulation for 6 h, the elevated release of nitrate, as the metabolite of NO, from BV-2 cells was detected as 27.0 ± 3.9 μM (1.6 ± 0.1-fold) as compared with the resting group of 17.1 ± 3.1 μM in the culture supernatants (). When carbamazepine (5, 10, and 20 μM) was added to BV-2 for 15 min before LPS stimulation, it decreased the level of nitrate in the culture supernatant at a concentration-dependent manner by 24.9 ± 3.4 μM (1.5 ± 0.1-fold), 21.0 ± 2.6 μM (1.3 ± 0.1-fold), and 19.4 ± 2.9 μM (1.2 ± 0.1-fold), respectively (n = 3). In contrast, NO release was inhibited by up to 75% at the concentration of 20 μM.
Figure 1. The effects of carbamazepine on NO release and iNOS production. (A) BV-2 cells (2 × 105 cells/ml) were dispensed on 12-well plates and treated with various concentrations of LPS (50, 100, 150, and 200 ng/ml) for 24 h. The cell lysates were obtained and analyzed for iNOS protein expression by Western blots. (B) For evaluation of nitrate production, BV-2 cells (1 × 106 cells/ml) were dispensed on 24-well plates and treated with the indicated concentrations of carbamazepine (5, 10, and 20 μM) for 15 min before treatment with LPS (150 ng/ml) for 6 h. Thereafter, cell-free supernatants were obtained and analyzed for nitrate production by assay kit, as detailed in “Methods” section. The data are shown as means ± S.E.M of three to four experiments. (C) For evaluation of iNOS protein expression, BV-2 cells were treated with various concentrations of carbamazepine (1, 5, 10, and 20 μM) for 15 min before treatment with LPS (150 ng/ml) for 24 h. The data are shown as means ± S.E.M of four to six experiments. ##p < 0.01, ###p < 0.001 as compared with the resting groups; *p < 0.05, ***p < 0.001 as compared with the vehicle under stimulation.
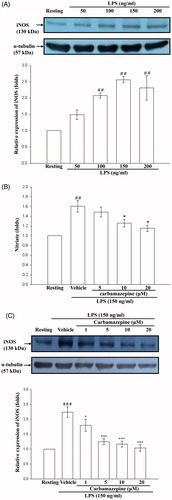
Consistently, it revealed that carbamazepine (1, 5, 10, and 20 μM) significantly inhibited LPS-stimulated the production of iNOS protein at a concentration-dependent manner by 1.8 ± 0.2-, 1.3 ± 0.1-, 1.2 ± 0.1-, and 1.0 ± 0.1-fold compared with 2.2 ± 0.2-fold of the vehicle group in BV-2 cells, respectively (n = 6, ). As shown in , LPS-induced iNOS expression was markedly attenuated by carbamazepine (20 μM) by up to approximately 96% inhibition.
The effect of carbamazepine on the expression of iNOS mRNA in BV-2 cells and cellular viability
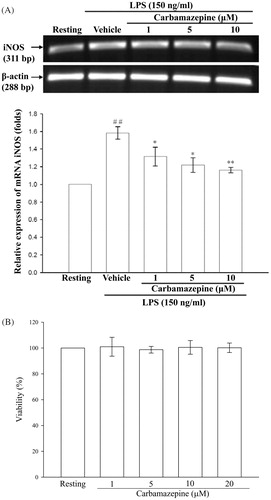
As shown in , LPS (150 ng/ml) markedly stimulated an increase in iNOS mRNA in BV-2 cells compared with the resting group. Moreover, pretreatment with carbamazepine (1, 5, and 10 μM) for 15 min significantly inhibited the LPS-induced iNOS mRNA expression. To further demonstrate whether carbamazepine interrupt the viability of BV-2 cells, the 24 h MTT assay was performed and showed that this agent had no effect on cellular viability (1 μM with 101.0 ± 7.3%, 5 μM with 98.7 ± 2.6%, 10 μM with 100.5 ± 5.3%, and 20 μM with 100.2 ± 3.7 viability, n = 3) ().
Figure 2. The effect of carbamazepine on the expression of iNOS mRNA in BV-2 cells and cellular viability. (A) BV-2 cells (5 × 105 cells/ml) were dispensed on 6-well plates and treated with different concentrations of carbamazepine (1, 5, and 10 μM) for 15 min before treatment with LPS (150 ng/ml) for 6 h. Following by extraction of total RNA and analysis of mRNA levels of iNOS and β-actin were performed by cDNA contents (311 bp and 288 bp) through the RT-PCR technique. The RT-PCR technique was performed as described in “Methods” section. The data were expressed and shown as the means ± S.E.M of three experiments. ##p < 0.01 as compared with the resting groups; *p < 0.05, **p < 0.01 as compared with the vehicle under stimulation. (B) Percentage of cellular MTT reduction was presented as the means ± S.E.M of three independent experiments.
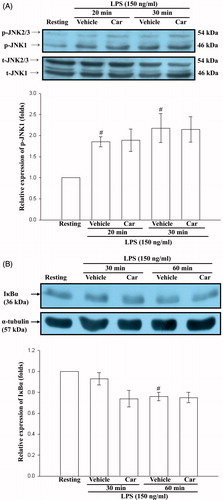
The effect of carbamazepine on JNK MAPK and NF-κB signaling
Because LPS is known to activate either JNK MAPK or NF-κB pathways, we next investigated whether the effect of carbamazepine on iNOS expression was mediated by these signaling pathways. The results showed that LPS treatment activated JNK MAPK and NF-κB signalings by Western blot analyses in our experimental condition (). However, carbamazepine (20 μM) had no significant effect on LPS-stimulated activation of either phosphorylation of JNK-1 or IκBα degradation at the indicated times, respectively (). These results suggest that the inhibitory effect of carbamazepine is not dependent on suppression of NF-κB or JNK MAPK signaling.
Figure 3. The effect of carbamazepine on JNK MAPK and NF-κB signaling. BV-2 cells (2 × 105 cells/ml) were dispensed on 12-well plates and treated with carbamazepine (Car) (20 μM) for 15 min before treatment with LPS (150 ng/ml) for the indicated times. Thereafter, the cell lysates were obtained and analyzed for phosphorylated (p-) JNK (A) and IκBα protein (B) expressions by Western blotting. The data are expressed as the means ± S.E.M of three to four experiments. #p < 0.05 as compared with the resting groups.
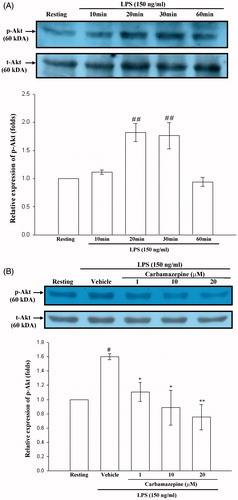
The effect of carbamazepine on Akt activation
Since Akt is also an important mediator of LPS-related signal pathway in microglial cells (Xing et al., Citation2008), we further investigated whether carbamazepine attenuates iNOS expression through interference with Akt signaling. As depicted in , a significant phosphorylation of Akt protein was observed in BV-2 cells during 20–30 min of treatment with LPS (150 ng/ml) by Western blot analyses. The results also indicated that LPS-induced phosphorylation of Akt was markedly inhibited by carbamazepine (1, 10, and 20 μM) by 1.11 ± 0.13-, 0.89 ± 0.25 -, and 0.75 ± 0.18-fold compared with 1.60 ± 0.04-fold of the vehicle group ().
Figure 4. The effect of carbamazepine on Akt activation. (A) BV-2 cells (2 × 105 cells/ml) were dispensed on 12-well plates and treated with the indicated concentrations of LPS (150 ng/ml) for 10, 20, 30, and 60 min. Then the cell lysates were obtained and analyzed for phospho-Akt protein expression by Western blots. (B) The same concentration of cells were dispended on 12-well plates and treated with different concentrations of carbamazepine (1, 10, and 20 μM) for 15 min before treatment with LPS (150 ng/ml) for 30 min. The cell lysates were also performed for phospho-Akt protein expression. The data are shown as the means ± S.E.M of three to four experiments. #p < 0.05, ##p < 0.01 as compared with the resting groups; *p < 0.05, **p < 0.01 as compared with the vehicle under stimulation.
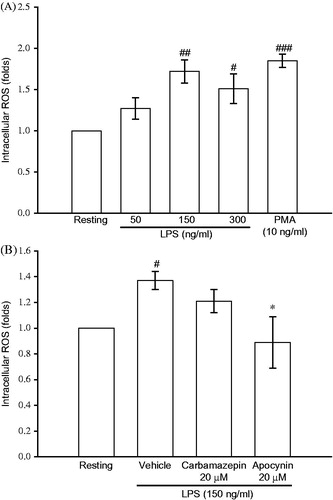
The effect of carbamazepine on ROS formation in activated BV-2 cells
In order to verify that the signal effectors are attenuated through ROS inhibition by carbamazepine in BV-2 cells, we studied the ROS formation by fluorescent probe assay. The fluorescent analysis shown in revealed that LPS (50, 150, and 300 ng/ml) exerted a concentration-dependent stimulation of ROS production in BV-2 cells. The ROS production was also triggered by a positive activator, PMA (10 nM), as 1.85 ± 0.08-fold compared with the resting condition (, n = 3). However, LPS (150 ng/ml)-induced ROS formation was not significantly affected by treatment with carbamazepine (20 μM) by 1.21 ± 0.09-fold compared with 1.37 ± 0.07-fold of the vehicle group (, n = 5), respectively. Although, a classical NADPH oxidase inhibitor apocynin at the same concentration exerted strong inhibition on the ROS formation ().
Figure 5. The effect of carbamazepine on ROS formation in activated BV-2 cells. BV-2 cells (2.5 × 105 cells/ml) were dispensed on 96-well plates and treated with DCFH-DA (20 μM) for 1 h. (A) Thereafter, the cells were activated with various concentrations of LPS (50, 150, and 300 ng/ml) or PMA (10 ng/ml) for 2 h. Then cellular fluorescence was analyzed for ROS production. (B) In contrast, the labeled cells were pretreated with either carbamazepine (20 μM) or apocynin (20 μM) for 15 min before activation by LPS (150 ng/ml) for the indicated time. The fluorescence was read immediately at wavelengths of 485 nm for excitation and 530 nm for emission on a fluorescence plate reader. The data are shown as mean ± S.E.M of three to five experiments. #p < 0.05, ##p < 0.01, ###p < 0.001 as compared with the resting groups; *p < 0.05 as compared with the vehicle under stimulation.
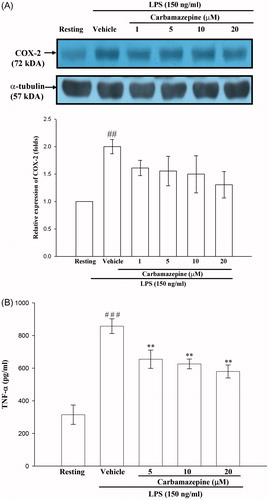
The effects of carbamazepine on cyclooxygenase-2 expression and TNF-α production
As shown in , LPS (150 ng/ml) exerted a marked stimulation of COX-2 protein synthesis and TNF-α production in BV-2 cells. Pretreatment with various concentrations of carbamazepine (1, 5, 10, and 20 μM) for 30 min before LPS had no significant effect on the expression of COX-2 by 1.6 ± 0.1-, 1.6 ± 0.3-, 1.5 ± 0.3-, and 1.3 ± 0.2-fold compared with 2.0 ± 0.1-fold of the vehicle group in BV-2 cells (). Likewise, the elevated level of TNF-α was partially affected by carbamazepine (5, 10, and 20 μM) by 655 ± 56, 626 ± 30, and 580 ± 40 pg/ml as compared with 857 ± 45 pg/ml of the vehicle group in activated BV-2 cells, respectively (n = 5, ). This indicates that carbamazepine affected the expression of a specific gene during stimulation of LPS.
Figure 6. The effects of carbamazepine on cyclooxygenase-2 expression and TNF-α production. BV-2 cells (2 × 105 cells/ml) were dispensed on 12-well plates and treated with different concentrations of carbamazepine (1, 5, 10, and 20 μM) for 15 min before treatment with LPS (150 ng/ml) for 24 h. (A) The cell lysates were obtained and analyzed for COX-2 protein expression by Western blotting. (B) For evaluation of TNF-α production, BV-2 cells (1 × 106 cells/ml) were dispensed on 24-well plates and treated with different concentrations of carbamazepine (5, 10, and 20 μM) for 15 min before treatment with LP supernatants were obtained and analyzed for TNF-α production by ELISA kit, as detailed in “Methods” section. The data are expressed as the means ± S.E.M of three experiments. ##p < 0.01, ###p < 0.001 as compared with the resting groups; **p < 0.01 as compared with the vehicle under stimulation.
Discussion
Activated microglial cells could damage neural cells by secreting a number of neurotoxic and proinflammatory mediators such as nitric oxide (NO) and prostaglandins (Block & Hong, Citation2005). The excessive production of NO by microglial iNOS has been found in the brain lesions of psychiatric and epileptic patients (Kamida et al., Citation2007). It is believed that NO exerts an important role in neuronal hyper-excitability evidenced in several epileptic disorders (Ferraro & Sardo, Citation2004). NO is also able to react rapidly with superoxide anion and produce peroxynitrite which mediates the oxidative and nitrating damage (Beckman et al., Citation1990).
It was reported that carbamazepine has a significant inhibitory effect on the inflammation-related complications (Iwamoto et al., Citation2011). Thus, we supposed that carbamazepine may affect microglial activation during neuroinflammatory processes. Our results showed that carbamazepine concentration dependently attenuated the production of NO and iNOS protein stimulated by LPS in microglial BV-2 cells. Consistently, the LPS-induced iNOS mRNA expression was also abrogated by carbamazepine in a concentration-dependent manner. According to these results, the inhibitory mechanism of carbamazepine could be ascribed to its inhibition on signal pathways.
NF-κB is an important transcription factor for the expression of brain inflammatory genes such as iNOS and COX-2 gene (Ridder & Schwaninger, Citation2009). Classically, it is well known that LPS stimulates NF-κB signaling through the activation of IκB kinase, and subsequent phosphorylation and degradation of IκBα in microglial cells (Watters et al., Citation2002). It was reported that inhibition of NF-κB could lead to down-regulation of iNOS expression (Lu et al., Citation2007). However, carbamazepine did not affect LPS-induced IκBα degradation in this study. These results suggested that carbamazepine attenuates LPS-induced iNOS expression through a mechanism which does not involve the inhibition of NF-κB activation in BV-2 microglial cells.
In contrast, MAPK and redox signalings may also play an important role in regulating the expression of iNOS gene in activated BV-2 cells (Hwang et al., Citation2004). In consistent with the previous report (Zhang et al., 2010), our results showed that JNK MAPK could be activated and phosphorylated under LPS stimulation. Pretreatment with either a dominant mutant JNK plasmid or a JNK inhibitor SP600125 could abrogate iNOS expression (Liu et al., 2010b). However, carbamazepine could not abrogate LPS-induced activation of JNK MAPK (). Additionally, the antioxidant such as N-acetylcystein could strongly down-regulate LPS-induced ROS formation, and thereby inhibit iNOS expression (Oh et al., Citation2009). However, ROS formation was not affected by carbamazepine in our experimental condition (). Collectively, neither MAPK nor redox signal was the major target of cabamazepine to reduce LPS-induced iNOS expression in the microglial cells.
Phosphatidylinositol 3-kinase (PI-3K) signaling and its downstream signaling molecule, Akt, are crucially implicated in the regulation of cellular mitogenesis, survival and inflammatory mediator expression (Brazil et al., Citation2004; Crowder & Freeman, Citation1998). It was also found the PI-3K/Akt signaling cascade plays a pivotal role in regulating the expression of iNOS gene in activated BV-2 cells (Hwang et al., Citation2004), and that Akt exerts active function after its specific regulatory domains being phosphorylated (Alessi et al., Citation1996). Our results were consistent with the previous findings (Liu et al., 2010b; Oh et al., Citation2009), Akt could be phosphorylated and activated by LPS stimulation in activated BV-2 cells. Furthermore, pretreatment with a pharmacological inhibitor of PI-3K (LY294002) could abrogate iNOS expression in LPS-activated microglial cells (Liu et al., 2010b). In addition, previous reports demonstrated that the bioactive eicosapentaenoic acid or TGF-β1 could exert attenuation of iNOS expression through Akt inhibition (Kim et al., Citation2004; Moon et al., Citation2007). Differently, resveratrol inhibited LPS-induced proinflammatory enzymes via an enhanced Akt-related mTOR-dependent pathway in BV-2 cells (Zhong et al., Citation2012). In contrast, it has been documented that cytokine-activated Akt could increase IκBα degradation or p65 translocation by phosphorylation of IKKα or Rel/p65 in HepG2 cells (Sizemore et al., Citation1999). Differentially, manipulation of Akt was not related to NF-κB inhibition in BV-2 cells by the addition of carbamazepine. Recently, carbamazepine was found to promote the acetylation of heat-shock protein 90 (Hsp 90) and disrupt its chaperone function (Meng et al., Citation2011). Therefore, Akt as a client protein of HSP 90 could be degraded after treatment of carbamazepine, and need to be proven. It was clearly shown in this study that carbamazepine significantly inhibited the LPS-induced phosphorylation and activation of Akt, but did not repress the NF-κB signaling. Therefore, these findings may imply that the LPS-activated Akt cascade is not upstream of NF-κB signaling in the microglial cells. Our results strongly indicated that PI-3K/Akt signaling pathway may play an important role in the anti-neuroinflammatory action of carbamazepine on the activated microglial cells.
Taken together, this study signified that carbamazepine attenuates LPS-induced iNOS expression predominantly by inhibiting PI-3K/Akt signaling pathway in activated BV-2 microglial cells. Given the down-regulation effect of carbamazepine on iNOS expression, this classical psychotropic agent should further be tested in animal models or clinical trials as a potential therapeutic agent for neuroinflammation.
Declaration of interest
The authors declare that there are no conflicts of interest. This work was financially supported by the research grants from the Cathay General Hospital (99-CGH-TMU-01-2) and National Science Council of Taiwan (NSC96-2320-B-038-021-MY3).
Related Research Data
References
- Alessi DR, Andjelkovic M, Caudwell B, et al. (1996). Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15:6541–51
- Al-lzki S, Pryce G, Hankey DJ, et al. (2014). Lesional-targeting of neuroprotection to the inflammatory penumbra in experimental multiple sclerosis. Brain 137:92–108
- Almgren M, Nyengaard JR, Persson B, Lavebratt C. (2008). Carbamazepine protects against neuronal hyperplasia and abnormal gene expression in the megencephaly mouse. Neurobiol Dis 32:364–76
- Beckman JS, Beckman TW, Chen J, et al. (1990). Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA 87:1620–4
- Bhat NR, Fan F. (2002). Adenovirus infection induces microglial activation: Involvement of mitogen-activated protein kinase pathways. Brain Res 948:93–101
- Block ML, Hong JS. (2005). Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog Neurobiol 76:77–98
- Brazil DP, Yang ZZ, Hemmings BA. (2004). Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci 29:233–42
- Brown GC, Bal-Price A. (2003). Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol Neurobiol 27:325–55
- Choi J, Nordli DR Jr, Alden TD, et al. (2009). Cellular injury and neuroinflammation in children with chronic intractable epilepsy. J Neuroinflamm 6:38–51
- Chou YC, Sheu JR, Chung CL, et al. (2010). Nuclear-targeted inhibition of NF-kappaB on MMP-9 production by N-2-(4-bromophenyl) ethyl caffeamide in human monocytic cells. Chem Biol Interact 184:403–12
- Chung CL, Sheu JR, Liu HE, et al. (2009). Dynaore, a dynamin inhibitor, induces PAI-1 expression in MeT-5A human pleural mesothelial cells. Am J Respir Cell Mol Biol 40:692–700
- Crowder RJ, Freeman RS. (1998). Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci 18:2933–43
- De Bragança AC, Moyses ZP, Magaldi AJ. (2010). Carbamazepine can induce kidney water absorption by increasing aquaporin 2 expression. Nephrol Dial Transplant 25:3840–5
- De Luca G, Di Giorgio RM, Macaione S, et al. (2006). Amino acid levels in some brain areas of inducible nitric oxide synthase knock out mouse (iNOS-/-) before and after pentylenetetrazole kindling. Pharmacol Biochem Behav 85:804–12
- De Sarro G, Gareri P, Falconi U, de Sarro A. (2000). 7-Nitroindazole potentiates the antiseizure activity of some anticonvulsants in DBA/2 mice. Eur J Pharmacol 394:275–88
- Ferraro G, Sardo P. (2004). Nitric oxide and brain hyperexcitability. In Vivo 18:357–66
- Himmerich H, Bartsch S, Hamer H, et al. (2013). Impact of mood stabilizers and antiepileptic drugs on cytokine production in-vitro. J Psychiatr Res 47:1751–9
- Hsiao G, Huang HY, Fong TH, et al. (2004). Inhibitory mechanisms of YC-1 and PMC in the induction of iNOS expression by lipoteichoic acid in RAW 264.7 macrophages. Biochem Pharmacol 67:1411–19
- Hwang SY, Jung JS, Lim SJ, et al. (2004). LY294002 inhibits interferon-gamma-stimulated inducible nitric oxide synthase expression in BV2 microglial cells. Biochem Biophys Res Commun 318:691–7
- Iwamoto T, Takasugi Y, Higashino H, et al. (2011). Antinociceptive action of carbamazepine on thermal hypersensitive pain at spinal level in a rat model of adjuvant-induced chronic inflammation. J Anesth 25:78–86
- Jin CY, Moon DO, Lee KJ, et al. (2006). Piceatannol attenuates lipopolysaccharide-induced NF-kappaB activation and NF-kappaB-related proinflammatory mediators in BV2 microglia. Pharmacol Res 54:461–7
- Johannessen LC. (2008). Antiepileptic drugs in non-epilepsy disorders: Relations between mechanisms of action and clinical efficacy. CNS Drugs 22:27–47
- Kamida T, Takeda Y, Fujiki M, et al. (2007). Nitric oxide synthase and NMDA receptor expressions in cavernoma tissues with epileptogenesis. Acta Neurol Scand 116:368–73
- Kim DH, Yoon BH, Jung WY, et al. (2010). Sinapic acid attenuates kainic acid-induced hippocampal neuronal damage in mice. Neuropharmacology 59:20–30
- Kim WK, Hwang SY, Oh ES, et al. (2004). TGF-beta1 represses activation and resultant death of microglia via inhibition of phosphatidylinositol 3-kinase activity. J Immunol 172:7015–23
- Lee K, Vivithanaporn P, Siemieniuk RA, et al. (2010). Clinical outcomes and immune benefits of anti-epileptic drug therapy in HIV/AIDS. BMC Neurol 10:44–55
- Lehnardt S, Massillon L, Follett P, et al. (2003). Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA 100:8514–19
- Liu HT, Du YG, He JL, et al. (2010b). Tetramethylpyrazine inhibits production of nitric oxide and inducible nitric oxide synthase in lipopolysaccharide-induced N9 microglial cells through blockade of MAPK and PI3K/Akt signaling pathways, and suppression of intracellular reactive oxygen species. J Ethnopharmacol 129:335–43
- Liu J, Wang A, Li L, et al. (2010a). Oxidative stress mediates hippocampal neuron death in rats after lithium-pilocarpine-induced status epilepticus. Seizure 19:165–72
- Lloyd E, Somera-Molina K, Van Eldik LJ, et al. (2008). Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small molecule improves long-term neurologic outcome in a mouse model of traumatic brain injury. J Neuroinflamm 5:28–41
- Lu DY, Tang CH, Liou HC, et al. (2007). YC-1 attenuates LPS-induced proinflammatory responses and activation of nuclear factor-kappaB in microglia. Br J Pharmacol 151:396–405
- Macdonald RL, Kelly KM. (1995). Antiepileptic drug mechanisms of action. Epilepsia 36:S2–12
- Mann L, Heldman E, Bersudsky Y, et al. (2009). Inhibition of specific adenylyl cyclase isoforms by lithium and carbamazepine, but not valproate, may be related to their antidepressant effect. Bipolar Disord 11:885–96
- Meng Q, Chen X, Sun L, et al. (2011). Carbamazepine promotes Her-2 protein degradation in breast cancer cells by modulating HDAC6 activity and acetylation of Hsp90. Mol Cell Biochem 348:165–71
- Moon DO, Kim KC, Jin CY, et al. (2007). Inhibitory effects of eicosapentaenoic acid on lipopolysaccharide-induced activation in BV2 microglia. Int Immunopharmacol 7:222–9
- Oh YT, Lee JY, Lee J, et al. (2009). Oleic acid reduces lipopolysaccharide-induced expression of iNOS and COX-2 in BV2 murine microglial cells: Possible involvement of reactive oxygen species, p38 MAPK, and IKK/NF-kappaB signaling pathways. Neurosci Lett 464:93–7
- Pawate S, Shen Q, Fan F, Bhat NR. (2004). Redox regulation of glial inflammatoryresponse to lipopolysaccharide and interferongamma. J Neurosci Res 77:540–51
- Pérez Martín JM, Fernández Freire P, Labrador V, Hazen MJ. (2008). Carbamazepine induces mitotic arrest in mammalian Vero cells. Mutat Res 637:124–33
- Phillips EJ, Mallal SA. (2009). HLA and drug-induced toxicity. Curr Opin Mol Ther 11:231–42
- Ribeiro LR, Fighera MR, Oliveira MS, et al. (2009). Methylmalonate-induced seizures are attenuated in inducible nitric oxide synthase knockout mice. Int J Dev Neurosci 27:157–63
- Ridder DA, Schwaninger M. (2009). NF-κB signaling in cerebral ischemia. Neuroscience 158:995–1006
- Siebel AM, Rico EP, Capiotti KM, et al. (2010). In vitro effects of antiepileptic drugs on acetylcholinesterase and ectonucleotidase activities in zebrafish (Danio rerio) brain. Toxicol In Vitro 24:1279–84
- Sizemore N, Leung S, Stark GR. (1999). Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the NF-kappaB p65/RelA subunit. Mol Cell Biol 19:4798–805
- Stepanovic-Petrovic RM, Tomic MA, Vuckovic SM, et al. (2011). Pharmacological interaction between oxcarbazepine and two COX inhibitors in a rat model of inflammatory hyperalgesia. Pharmacol Biochem Behav 97:611–18
- Stoll G, Jander S. (1999). The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol 58:233–47
- Trimble M. (2002). Carbamazepine, clinical efficacy and use in psychiatric disorders. In: Levy RH, Mattson RH, Meldrum BS, Perucca E, eds. Antiepileptic Drugs. Philadelphia: Lippincott Williams & Wilkins, 278–84
- Vezzani A, Granata T. (2005). Brain inflammation in epilepsy: Experimental and clinical evidence. Epilepsia 46:1724–43
- Von Borstel Smith M, Crofoot K, Rodriguez-Proteau R, Filtz TM. (2007). Effects of phenytoin and carbamazepine on calcium transport in Caco-2 cells. Toxicol In Vitro 21:855–62
- Wang J. (2010). Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol 92:463–77
- Watters JJ, Sommer JA, Pfeiffer ZA, et al. (2002). A differential role for the mitogen-activated protein kinases in lipopolysaccharide signaling: The MEK/ERK pathway is not essential for nitric oxide and interleukin 1beta production. J Biol Chem 277:9077–87
- Willhauck MJ, O Kane DJ, Wunderlich N, et al. (2010). Stimulation of retinoic acid-induced functional sodium iodide symporter (NIS) expression and cytotoxicity of 131I by carbamazepine in breast cancer cells. Breast Cancer Res Treat 125:377–86
- Xing B, Xin T, Hunter RL, Bing G. (2008). Pioglitazone inhibition of lipopolysaccharide-induced nitric oxide synthase is associated with altered activity of p38 MAP kinase and PI3K/Akt. J Neuroinflamm 5:4–14
- Zhang L, Wu C, Zhao S, et al. (2010). Demethoxycurcumin, a natural derivative of curcumin attenuates LPS-induced pro-inflammatory responses through down-regulation of intracellular ROS-related MAPK/NF-kappaB signaling pathways in N9 microglia induced by lipopolysaccharide. Int Immunopharmacol 10:331–8
- Zhong LM, Zong Y, Sun L, et al. (2012). Resveratrol inhibits inflammatory responses via the mammalian target of rapamycin signaling pathway in cultured LPS-stimulated microglial cells. PLoS One 7:1–12