
Abstract
Context: Cardiac cell death and fatal arrhythmias during myocardial ischemia/reperfusion (I/R) can be reduced by p38 MAPK inhibition. However, the effects of p38 MAPK inhibition on cardiac mitochondria have not been investigated.
Objective: We tested the hypothesis that p38 MAPK inhibition at different times during I/R protects cardiac mitochondrial functions.
Materials and methods: Adult Wistar rats were subjected to 30 min of left anterior descending coronary artery (LAD) occlusion, followed by 120 min of reperfusion. A 2 mg/kg bolus infusion of p38 MAPK inhibitor, SB203580, was given before or during ischemia, or at reperfusion. Mitochondrial function and ultrastructure were assessed and Western blots were performed.
Results: Administration of SB203580 at any time point of I/R significantly attenuated the mitochondrial ultrastructure change, mitochondrial swelling, by increasing the absorbance at 540 nm (I/R control 0.42 ± 0.03; pretreatment 0.58 ± 0.04; during ischemia 0.49 ± 0.02; at reperfusion 0.51 ± 0.02, p < 0.05), similar to reactive oxygen species (ROS) generation (I/R control 1300 ± 48; pretreatment 1150 ± 30; during ischemia 1000 ± 50; at reperfusion 1050 ± 55, p < 0.05). Only SB203580 given before or during ischemia attenuated mitochondrial membrane depolarization (I/R control 0.78 ± 0.04; pretreatment 1.02 ± 0.03; during ischemia 1.05 ± 0.12, p < 0.05). In addition, pre-treatment of SB203580 significantly reduced the phosphorylation of p53, CREB, Bax, cytochrome c, and cleaved caspase 3.
Discussion and conclusion: The results from this study showed for the first time that p38 MAPK inhibition protects mitochondria from I/R injury.
Introduction
Acute myocardial infarction has been a serious health burden in most countries around the world for many decades (World Health Organization [WHO], Citation2008). Although myocardial reperfusion has been shown to be an effective way of myocardial salvage, reperfusion itself has been known to cause cellular injury (Gottlieb et al., Citation1994). One of the major underlying cellular mechanisms following myocardial ischemia/reperfusion (I/R) injury has been shown to be cardiac mitochondrial dysfunction. For example, I/R injury has been shown to cause cardiac mitochondrial swelling, increased cardiac mitochondrial reactive oxygen species (ROS) production, and cardiac mitochondrial membrane depolarization (Baines, Citation2009). Cardiac mitochondrial dysfunction following I/R injury is known to trigger cellular necro-apoptosis (Gottlieb, Citation2011).
Previous studies demonstrated that the activation of the 38 kDa mitogen which activates protein kinase (p38 MAPK) during myocardial ischemia aggravates lethal cellular injury, particularly during I/R injury (Bogoyevitch et al., Citation1996; Clark et al., Citation2007; Kumphune et al., Citation2012). The pharmacological catalytic site inhibitor SB203580 has been shown to prevent p38 MAPK activation, and has beneficial effects on the heart, including reduced cardiomyocyte death and infarct size as well as attenuation of left ventricular function impairment in the myocardial I/R model (Barancik et al., Citation2000; Clark et al., Citation2007; Kumphune et al., Citation2010; Surinkaew et al., Citation2013). Recently, we reported the cardioprotective effects of SB203580 administered at different time courses of I/R injury models, and demonstrated that SB203580 given at each time course could reduce the infarct size even when given at the reperfusion onset (Surinkaew et al., Citation2013). Interestingly, SB203580 not only reduced the infarct size but also decreased I/R-induced arrhythmias. The anti-arrhythmic effect of SB203580 was clearly seen only when the drug was given prior to reperfusion. These findings indicated the importance of the timing of drug administration on the therapeutic outcomes. Since cardiac mitochondria have been shown to play an important role in arrhythmogenesis and infarct size during I/R, it is proposed that the beneficial effect of p38 MAPK inhibitor in infarct size reduction and arrhythmias could possibly be due to its effect on the cardiac mitochondria. However, this hypothesis has never been investigated.
In addition, it has been known that the activation of p38 MAPK could activate several downstream signaling molecules such as p53 (Perfettini et al., Citation2005; Singhal et al., Citation2002; Thornton & Rincon, Citation2009), cAMP response element-binding protein (CREB) (Arnould et al., Citation2002; Lochner et al., Citation2009). These signaling molecules play an important role in controlling the cellular necro-apoptotic regulatory molecules such as Bax, Bcl2, caspase 3, and cytochrome c, possibly via the mitochondrial apoptotic pathway (Krijnen et al., Citation2002; Lundberg & Szweda, Citation2004; Murphy et al., Citation2000; Saikumar et al., Citation1998). However, the effects of SB203580 on these signaling markers, in terms of linkage between cardiac mitochondrial dysfunction and cell death, have not been intensively investigated. Therefore, in the present study, we determined the effects of p38 MAPK inhibitor, SB203580, given at different times during the I/R cycle on cardiac mitochondrial function, including mitochondrial swelling, mitochondrial ROS production, mitochondrial membrane potential changes (ΔΨm), as well as cardiac mitochondrial ultrastructure, in an in vivo I/R rat model. Moreover, the signaling cascades involved in cardiac mitochondrial protection downstream to p38 MAPK activation including p53 and CREB as well as the necro-apoptotic regulatory molecules including Bax, Bcl2, caspase 3, and cytochrome c were also determined.
Materials and methods
Animal model
Adult male Wistar rats weighing 350–400 g were obtained from the National Animal Center, Salaya Campus, Mahidol University, Bangkok, Thailand. All animals were housed in a room maintained under constant environmental conditions (temperature 22–25 °C and a constant 12 h light/dark cycle) and received a standard rat diet and water ad libitum. All animal experiments were approved by the Institutional Animal Care and Use Committees, and conformed to the Guide for the Care and Use of Animals of the Faculty of Medicine, Chiang Mai University, Chiang Mai, Thailand.
Ischemia and reperfusion protocol
Myocardial ischemia/reperfusion (I/R) was induced by the left anterior descending coronary artery (LAD) ligation for 30 min, followed by reperfusion for 120 min as described previously (Lv et al., Citation2008). An ischemic heart was confirmed by the change in the myocardial tissue color and ST elevation on the electrocardiogram that was recorded throughout the experiment. The left femoral vein was cannulated for administration of either 2 mg/kg SB203580HCl (Tocris, Ellisville, MO) (Ward et al., Citation2001) or normal saline as a vehicle at 0.33 ml/min for 3 min (Lv et al., Citation2008). In the treatment groups, rats were divided into three treatment groups to receive one of the following; SB203580 at 15 min before LAD occlusion (pretreatment group), SB203580 at 15 min after the onset of ischemia (during ischemia group), or SB203580 at the onset of reperfusion (reperfusion group) (). At the end of the reperfusion period, the heart was excised and washed in cold saline solution. The whole ventricular tissue was homogenized for mitochondrial study or Western blot analysis.
Figure 1. Study protocol for experimental groups and timing of SB203580 (SB) or normal saline solution (NSS) administration.
Mitochondrial isolation
Mitochondria were freshly isolated from myocardial tissue by differential centrifugation as describe previously (Thummasorn et al., Citation2011). Briefly, ventricular tissues were homogenized in ice-cold isolated buffer (300 mM sucrose, 0.2 mM EGTA, 5 mM TES, pH 7.2) and centrifuged at 800 × g, 4 °C for 5 min. Then, the supernatants were collected and re-centrifuged at 8800 × g, 4 °C for 5 min. The mitochondrial pellet was washed by resuspending in ice-cold isolation buffer and re-centrifuged at 8800 × g, 4 °C for 5 min. The isolated mitochondrial and cytosolic fraction was subjected to Western blot analysis using rabbit polyclonal antibody against VDAC1/Porin – Mitochondrial loading control (Abcam, Cambridge, MA).
The mitochondrial protein concentration was determined by the bicinchoninic acid (BCA) method, using bovine serum albumin (BSA) as a standard. A similar concentration of isolated cardiac mitochondria at 0.4 mg/ml was used to examine mitochondrial swelling, mitochondrial ROS production, and alteration of mitochondrial membrane potential (ΔΨm).
Determination of mitochondrial swelling
To determine the sensitivity to membrane permeability transition (mPT), the mitochondrial swelling was measured. The isolated cardiac mitochondria were re-suspended in respiration buffer consisting of 100 mM KCl, 50 mM sucrose, 10 mM HEPES, and 5 mM KH2PO4. The permeability transition-induced swelling of mitochondria was measured by rapid loss of the absorbance at λ 540 nm by the spectrophotometric method. The isolated cardiac mitochondria (0.4 mg/ml) were incubated with 1.5 ml of respiration buffer, and then the decrease in the absorbance was measured for 30 min at room temperature. The data were represented in arbitrary units of absorbance. The decreased absorbance indicated mitochondrial swelling (Thummasorn et al., Citation2011).
Determination of mitochondrial ROS production
The mitochondrial ROS production was assessed by measuring the intensity of the fluorescent signal of fluorescent 2′,7′-dichlorohydrofluorescein (DCF), which was converted from non-fluorescent 2′,7′-dichlorofluorescein-diacetate (DCFH-DA) in the presence of ROS (Li et al., Citation2001). Isolated cardiac mitochondria (0.4 mg/ml) were incubated with 2 µM DCFH-DA for 30 min at room temperature. The fluorescence intensity was determined by a fluorescence microplate ready with the excitation at λ 485 nm and emission at λ 530 nm. The ROS level was expressed in arbitrary units of fluorescence intensity of DCF. The increased fluorescent intensity indicated increased ROS production (Thummasorn et al., Citation2011).
Determination of mitochondrial membrane potential changes
To test the hypothesis that treatment by SB203580 can protect the loss of ΔΨm, the JC1 dye was used to measure ΔΨm of cardiac mitochondria function. The JC-1 or 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide is a lipophilic cationic dye that is capable of entering the mitochondrial membrane. The monomeric form of JC-1 produces a green fluorescence. Increasing the mitochondrial membrane potential changes (ΔΨm) caused aggregation of the dye, which appeared as red fluorescein. Alteration of ΔΨm causes changes in the red:green ratio. Isolated cardiac mitochondria (0.4 mg/ml) were incubated with 5 µM JC-1 at 37 °C for 30 min. The fluorescence intensity for monomeric green fluorescein was determined by a fluorescence microplate reader with the excitation at λ 485 nm and emission at λ 530 nm, while the aggregate red fluorescein was determined by fluorescence microplate reader with the excitation at λ 485 nm and emission at λ 590 nm. The ratio of red to green fluorescence intensity ratio was determined. The decreased red to green fluorescent intensity ratio indicated mitochondrial membrane depolarization (Thummasorn et al., Citation2011).
Identification of cardiac mitochondrial ultrastructure
The mitochondrial pellet was fixed in 2.5% glutaraldehyde at 4 °C overnight. Then, the pellet was rinsed in 0.1 M phosphate buffer (PO4) for 15 min twice and post-fixed in 1% cacodylate-buffer osmium tetroxide for 2 h at room temperature. The mitochondrial pellet was rinsed in 0.1 M phosphate buffer (PO4) 5 min twice and was dehydrated in a graded series of ethanol, 50% ethanol for 5 min twice, 70% ethanol for 5 min twice, 85% ethanol for 5 min twice, 95% ethanol for 5 min twice, and 100% ethanol for 5 min twice. After that, the pellet was infiltrated with propylene oxide (PO) for 10 min twice, followed by the cocktail between resin and PO in a 1:2 ratio for 30 min, resin and PO in a 1:1 ratio for 60 min and resin overnight, respectively. On the next day, the pellet was embedded in EM-bed 812 resin (Thummasorn et al., Citation2011). Ultrathin sections were cut with diamond knife, placed in copper grids, and stained with uranyl acetate and lead citrate. The cardiac mitochondria were identified with a transmission electron microscope (TEM) (Thummasorn et al., Citation2011).
Western blot analysis
The heart tissue was homogenated in extraction buffer (20 mmol/L Tris HCl, 1 mmol/L Na3VO4, 5 mmol/L NaF). The heart protein was collected and subjected to 10% or 15% SDS-polyacrylamide gel electrophoresis then transferred to polyvinylidene difluoride membranes, which were blocked for 2 h with 5% non-fat milk in Tris-buffered saline (pH 7.4) containing 0.1% Triton X-100. The membranes were probed overnight at 4 °C with the appropriate primary antibody as follows: total-p38, phospho-p38, and phospho-HSP27 (Cell Signaling Technology, Danvers, MA), total-p53, phospho-p53, total-CREB, phospho-CREB, Bax, Bcl2, Cytochrome c, caspase-3 (Santa Cruz Biotechnology, Inc, Santa Cruz, CA). After washing and exposure to horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature, antibody–antigen complexes were visualized by enhanced chemiluminescence assay. Bands corresponding to the protein of interest appeared as dark regions on the developed film. The film images of the Western blots were scanned and were analyzed using Image J (NIH image, National Institutes of Health, Bethesda, MD) analysis software (Tanno et al., Citation2003). For quantitation of the proteins of interest, phosphorylated proteins were normalized to total protein expression.
Statistical analysis
All data were expressed as mean ± SEM. Statistical analysis was performed with one-way analysis of variance (ANOVA) and post-hoc analysis with LSD. All procedures were performed with an SPSS statistical program (Version 15.0, SPSS Inc., Chicago, IL). A p-values < 0.05 was accepted as statistically significant.
Results
p38 MAPK inhibition by SB203580 attenuated cardiac mitochondrial dysfunction following I/R injury
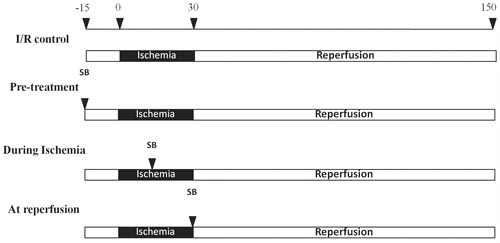
Isolated cardiac mitochondria used in this study were subjected to Western blot analysis against Anti-VDAC1/porin protein to ensure that the isolated protein contain mitochondria. The results showed that VDAC1/porin protein was detected in isolated mitochondrial fraction, but not in cytosolic fraction (). The myocardial I/R injury caused cardiac mitochondrial swelling by decreased absorbance at 540 nm, increased mitochondrial ROS production, and mitochondrial membrane depolarization (). Although administration of SB203580 at any time point of I/R protocol significantly attenuated cardiac mitochondrial swelling (), pretreatment of SB203580 was found to be the most effective timing to prevent cardiac mitochondrial swelling (pretreatment 0.58 ± 0.04 versus I/R control 0.42 ± 0.03, p < 0.05), compared with drug treatment during ischemia (during ischemia 0.49 ± 0.02 versus I/R control 0.42 ± 0.03, p < 0.05) or at the onset of reperfusion (at reperfusion 0.51 ± 0.02 versus I/R control 0.42 ± 0.03, p < 0.05) (). Administration of SB203580 also significantly reduced ROS production in cardiac mitochondria caused by I/R injury in all groups, compared with the I/R control group (I/R control 1300 ± 48; pretreatment 1150 ± 30; during ischemia 1000 ± 50; at reperfusion 1050 ± 55, p < 0.05) (). For cardiac mitochondrial membrane potential alteration, we found that administration of SB203580 prior to ischemia or during ischemia significantly prevented the change of mitochondrial membrane potential caused by I/R injury, when compared with the I/R control group (I/R control 0.78 ± 0.04; pretreatment 1.02 ± 0.03; during ischemia 1.05 ± 0.12) (). However, SB203580 administered at the onset of reperfusion failed to prevent mitochondrial depolarization (I/R control 0.78 ± 0.04; at reperfusion 0.71 ± 0.05) ().
Figure 2. Effects of SB203580 on cardiac mitochondrial function. (A) Western blot analysis using antibody against mitochondrial VDAC1/Porin in cytoplasmic fraction (C), and mitochondrial fraction (M) of isolated cardiac mitochondria. (B) Mitochondrial swelling, (C) mitochondrial ROS production, (D) mitochondrial membrane potential change, when SB203580 was administered before, during ischemia, or at the onset of reperfusion in ischemia/reperfusion rats (n = 4–7 animals/group). *p < 0.05 versus the vehicle group, #p < 0.05 versus the pretreatment group.
Inhibition of p38 MAPK protected I/R-induced cardiac mitochondrial ultrastructure disruption
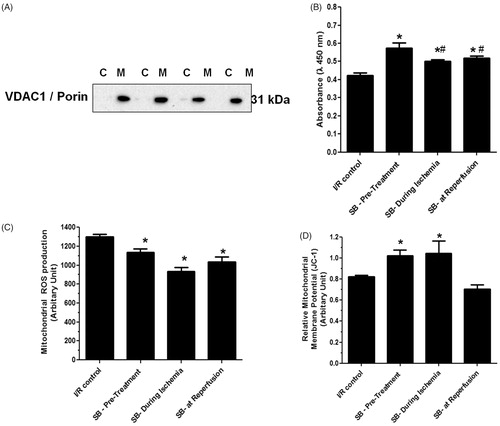
Our results demonstrated that I/R injury not only caused cardiac mitochondrial dysfunction but also distorted the cardiac mitochondrial ultrastructure by increasing matrix space and disorganization of the cristae (). Administration of SB203580 at any time points could preserve the cardiac mitochondrial ultrastructure from the disruption caused by I/R injury (). Interestingly, administration of SB203580 prior to ischemia gave the most effective treatment to protect I/R induced-cardiac mitochondria ultrastructure rupture (), when compared with the groups received SB203580 during ischemia or at the onset of reperfusion.
Figure 3. Effects of SB203580 on cardiac mitochondrial ultrastructure. Cardiac mitochondria were isolated from rats subjected to myocardial ischemia/reperfusion injury (A) in the presence of SB203580 before (B), during ischemia (C), or at the onset of reperfusion (D). The mitochondrial pellet was embedded in EM-bed 812 resin. Ultrathin sections were cut and stained with uranyl acetate and lead citrate. *p < 0.05 versus the vehicle group (n = 4–5 animals/group).
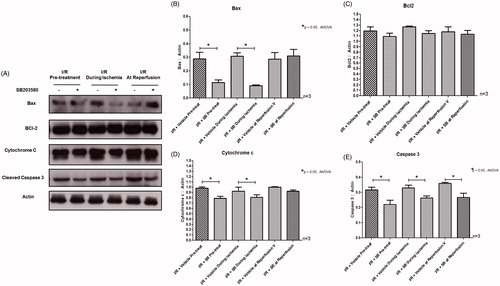
p38 MAPK inhibitor, SB203580, protected cardiac mitochondria by attenuation of apoptotic regulatory molecules activation
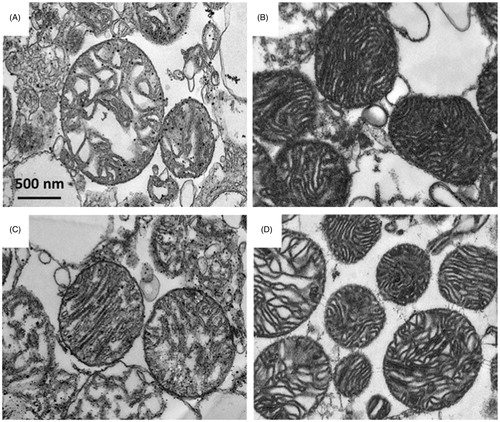
Myocardial I/R has been shown to cause activation of p38 MAPK and its activities by phosphorylation of downstream signaling molecules HSP27 (Larsen et al., Citation1997). The administration of the p38 MAPK inhibitor SB203580, prior to or during ischemia, significantly reduced p38 MAPK phosphorylation as well as its activity to phosphorylate the downstream substrate HSP27 (). However, SB203580 given at the onset of reperfusion did not reduce the p38 MAPK phosphorylation as well as the level of phosphorylated HSP27 (). Moreover, we found that pretreatment of SB203580 inhibited the phosphorylation of p53 and CREB, while SB203580 given during ischemia failed to inhibit the activation of these two downstream molecules (). However, SB203580 given at the onset of reperfusion inhibited the phosphorylation of p53 and CREB (). Since mitochondrial dysfunction is the key determination for cell death, especially the necro-apoptosis in I/R injury (Gottlieb, Citation2011), the effects of SB203580 on apoptotic regulatory molecules such as Bax, Bcl2, caspase 3, and cytochrome c were also determined. Our results showed that administration of SB203580 at every time point reduced the level of cleaved caspase 3 (), although SB203580 given prior to or during ischemia, but not at the onset of reperfusion, significantly decreased Bax expression and cytochrome c level (), without any changes in levels of Bcl2 expression ().
Figure 4. (A) Effects of SB203580 on p38 MAPK activation and downstream substrates, HSP27, p53, and CREB. The heart homogenates were collected and subjected to Western blot analysis to detect the activation of interested proteins. (B–E) The quantitation of fold phosphorylation of p38 MAPK activation and downstream substrates, HSP27, p53, and CREB by Western blot analysis (n = 3 animals/group). *p < 0.05 versus the vehicle group.
Figure 5. (A) Effects of SB203580 on apoptotic regulatory proteins. The heart homogenates were collected and subjected to Western blot analysis to detect the expression of Bax, Bcl2, Cytochrome c, and cleaved caspase-3. (B–E) The quantitation of protein expression of Bax, Bcl2, Cytochrome c, and cleaved caspase-3 by Western blot analysis (n = 3 animals/group). *p < 0.05 versus the vehicle group.
Discussion
Cardiac mitochondrial dysfunction is known as a key mechanism involved in I/R injury and cardiac cell death (Baines, Citation2009; Baines et al., Citation2007; Berkich et al., Citation2003; Di Lisa et al., Citation2007; Halrestrap et al., Citation2004; Jennings & Ganote, Citation1976). The excessive formation of ROS during I/R injury has been shown to induce the prolonged-opening of mitochondria permeability transition pore (MPTP), thus dissipating the proton electrochemical gradient or ΔΨm, consequently causing ATP insufficiency, leading to further ROS generation and the loss of intact cardiac mitochondrial ultrastructure, which finally resulted in cardiac mitochondrial swelling and rupture (Vaseva et al., Citation2012). This process is known to trigger the apoptotic program due to the leakage of pro-apoptotic molecules from ruptured mitochondria (Green & Reed, Citation1998). Therefore, prevention or attenuation of the degree of cardiac mitochondrial dysfunction caused by I/R injury is one of the most promising clinical targets.
In the present study, we demonstrated for the first time the protective effect of p38 MAPK inhibitor, SB203580, on cardiac mitochondrial function in the heart underwent I/R injury, and provided molecular mechanistic insights of cardiac mitochondrial protection by p38 MAPK inhibiting necro-apoptosis regulatory cascades. The major findings of this study were that in the heart that underwent I/R injury, administration of SB203580 before or during ischemia attenuated cardiac mitochondrial dysfunction caused by I/R as indicated by preventing mitochondrial swelling, reducing mitochondrial ROS generation, and attenuating mitochondrial membrane potential depolarization. However, giving SB203580 at the onset of reperfusion failed to prevent the loss of mitochondrial membrane potential, in which cardiac mitochondrial depolarization plays an important role in arrhythmogenesis (O'Rouke, Citation2000). These findings could provide the mechanistic explanation of our previous report, which demonstrated from our previous study that the infarct size reduction was found at all-time points of SB203580 treatment, but treatment of SB203580 before or during ischemia, but not at reperfusion, decreased the incidence of ventricular tachycardia/ventricular fibrillation (VT/VF) incidence (Surinkaew et al., Citation2013). Although SB203580 showed a protective effect on cardiac mitochondrial swelling, ROS production, and mitochondrial membrane potential depolarization, the mitochondrial respiration was not performed and was considered a limitation of this study.
In response to I/R stress, p53 protein has been known to be activated and accumulated in the mitochondrial matrix, induced ROS generation and subsequently caused transient mitochondrial outer membrane permeabilization (MOMP), resulting in the release of cytochrome c, which subsequently cause apoptosis by activation of caspase 3 (Vaseva et al., Citation2012). Therefore, p53 activation in I/R is considered as a key molecule to trigger and regulate mitochondrial cellular necro-apoptosis (Krijnen et al., Citation2002). It is important to note that p53 could be phosphorylated by p38 MAPK (Bulavin et al., Citation1999; Perfettini et al., Citation2005; Thornton & Rincon, Citation2009), and mediated cell death (Singhal et al., Citation2002), suggesting that the significant linkage of p38 MAPK and p53 regulates mitochondrial apoptotic pathway. In the present study, administration of p38 MAPK inhibitor prior to LAD ligation significantly reduced p53 phosphorylation as well as significantly decreased Bax expression, cytochrome c, and the level of cleaved caspase 3. These molecular findings may explain the effect of p38 MAPK inhibition prior to the ischemic period significantly attenuated I/R-induced cardiac mitochondrial dysfunction and also decreased apoptotic cascade activation. This suggested that the treatment of p38 MAPK inhibitor before an ischemic insult is the effective timing of treatment in terms of cardiac mitochondrial protection and reduction of apoptotic signaling activation. Similar to the report from Kimura et al. (Citation2006) that pretreatment of p38 MAPK inhibitor prior to myocardial ischemia could inhibit mitochondrial morphological damage. However, this study lacked information regarding mitochondrial function. Despite these facts, clinical application is limited since most acute myocardial infarction patients already have coronary occlusion prior to their arriving at the hospital. Therefore, administration of p38 MAPK inhibitor during myocardial ischemia or in reperfusion period would be more appropriate in an actual clinical setting.
Inhibition of p38 MAPK during ischemia or at the onset of reperfusion also provided cardioprotective benefits, but at a lower degree of protection compared to the pre-treatment. Administration of SB203580 during ischemia reduced p38 MAPK activity without affecting the downstream molecules p53. Since p53 has been shown to be rapidly phosphorylated within 5 min after onset of ischemia (Kim et al., Citation2013), therefore, administration of SB203580 15 min after the onset of ischemia was not early enough to inhibit p38 MAPK induced p53 phosphorylation. However, giving SB203580 during ischemia reduced the expression of pro-apoptotic protein Bax, cleaved caspase 3, which could attenuate cell death. Therefore, giving SB203580 late after the onset of ischemia may not be capable of deactivating p53 molecules that have been rapidly phosphorylated in the ischemic episode, but could inhibit the signaling cascade that sufficiently attenuates mitochondrial damage and cell death.
Although administration of SB203580 at the onset of reperfusion could not protect mitochondria membrane depolarization, it was still sufficient to attenuate cardiac mitochondrial swelling, ROS production, and mitochondrial structure, although to a lesser extent when compared with giving SB203580 before or during ischemia. Administration of SB203580 at the onset of reperfusion failed to reduce p38 MAPK activity on HSP27, but decreased the phosphorylation of p53 and CREB. These findings suggest that p53 and CREB are downstream molecules of p38 MAPK that were not activated via HSP27, or could possibly be regulated by other unidentified signaling pathways that could be affected by SB203580. Furthermore, SB203580 given at reperfusion could only reduce cleaved caspase 3 levels without any change in the Bax, Bcl2, and cytochrome c. The reduction of cleaved caspase 3 level could sufficiently attenuate the apoptotic pathway, which is consistent with the results from our previous report that SB203580 given at the onset of reperfusion also reduced the infarct size (Surinkaew et al., Citation2013).
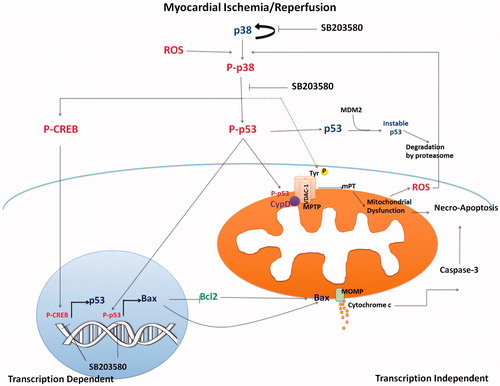
Inhibition of p38 MAPK by SB203580 reduced the p53 activation and Bax expression level, which may in turn attenuate the release of cytochrome c level to trigger the activity of caspase 3 mediated apoptosis (Hsu et al., Citation2007; Perfettini et al., Citation2005; Son et al., Citation2009). The impairment of mitochondrial activity has been found to activate cAMP-responsive element-binding protein (CREB) phosphorylation at Ser133 (Arnould et al., Citation2002), which in turn activated p53 in a transcriptional dependent manner (). Down regulation of p53 could also affect the p53-mediated mitochondrial dysfunction and apoptosis. Therefore, inhibition of CREB phosphorylation could target a p53 regulation. It has been shown that, during ischemia, CREB is activated as a downstream event of p38 MAPK activation (Lochner et al., Citation2009), in which its phosphorylation could be inhibited by SB203580. On the contrary, we found that the protein expression level of p53 was not changed, suggesting a transcriptional-independent mechanism. This could be due to the study protocol that aimed to study the acute effects of the p38 MAPK inhibitor. The duration of inhibitor treatment as well as duration of I/R protocol may not be sufficient to initiate the transcriptional processes of p53. In this study, administration of SB203580 inhibited p53 phosphorylation and subsequently influenced Bax expression, resulting in decreased mitochondrial membrane permeability, cytochrome c, and caspase 3 levels (). The limitation of this study was that our results provided limited mechanistic insights of SB203580 on cardiac mitochondria, since the protein levels of apoptotic factors, e.g., Bax, Bck-2, and Cytochrome c as well as p53 and CREB were performed in homogenated whole heart tissue. This may not sufficiently provide the information regarding the translocation of these apoptotic factors from cytoplasm to mitochondria. In addition, phosphorylation of the voltage-dependent anion channel (VDAC) -1, which is a porin protein involved in mitochondrial regulator of cell survival (Baines et al., Citation2007; Cesura et al., Citation2003; McEnery et al., Citation1992; Premkumar & Simantov, Citation2002), facilitates other protein binding in MPTP and mediates mitochondrial damage. Schwertz et al. (Citation2007) reported that VDAC-1 was a downstream substrate of p38 MAPK during I/R injury. Inhibition of p38 MAPK by PD169316 has been shown to significantly reduce the phosphorylation of VDAC-1, and reduce cardiac cell injury. However, the effects of SB203580 on VDAC-1 phosphorylation in cardiac mitochondria from myocardial I/R model will need to be further investigated.
Figure 6. The proposed mechanistic pathway of I/R-activated p38 MAPK and downstream activation involving mitochondrial trigger cell death. Myocardial I/R injury caused p38 MAPK activation, which consequently activated p53 phosphorylation. Phosphorylated p53 stabilized and accumulated in mitochondrial matrix during I/R injury and mediated MPTP opening. Activation of p53 also activated proapoptotic Bax protein, which regulated cytochrome c release, and activation of caspase 3 (transcription independent manner). p38 MAPK also phosphorylated CREB, which in turn regulated p53 gene transcription. Inhibition of p38 MAPK by SB203580 reduced p53 phosphorylation, CREB phosphorylation, and thus protected cardiac mitochondria from I/R injury and cell death.
Conclusions
In summary, this is the first report demonstrating that p38 MAPK inhibition by SB203580 reduced cardiac mitochondrial dysfunction caused by I/R injury through the attenuation of the mitochondrial apoptotic pathway. Moreover, our data suggests that the therapeutic potential of SB203580 in protecting cardiac mitochondria from I/R injury provides more clinical benefit when given prior to reperfusion.
Declaration of interest
The authors report that they have no conflicts of interest. This study was supported by grants from the Thailand Research Fund MRG5480017 (SK), the Royal Golden Jubilee Ph.D. Program (SS, NC), Thailand Research Fund RTA5580006 (NC), BRG5780016 (SC), the Chiang Mai University Center of Excellence Award (NC) and a NSTDA Research Chair Grant from the National Science and Technology Development Agency (NC).
Related Research Data
References
- Arnould T, Vankoningsloo S, Renard P, et al. (2002). CREB activation induced by mitochondrial dysfunction is a new signaling pathway that impairs cell proliferation. EMBO J 21:53–63
- Baines CP. (2009). The mitochondrial permeability transition pore and ischemia–reperfusion injury. Basic Res Cardiol 104:181–8
- Baines CP, Kaiser RA, Sheiko T, et al. (2007). Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 9:550–5
- Barancik M, Htun P, Strohm C, et al. (2000). Inhibition of the cardiac p38-MAPK pathway by SB203580 delays ischemic cell death. J Cardiovasc Pharmacol 35:474–83
- Berkich DA, Salama G, Lanoue KF. (2003). Mitochondrial membrane potentials in ischemic hearts. Arch Biochem Biophys 420:279–86
- Bogoyevitch MA, Gillespie BJ, Ketterman AJ, et al. (1996). Stimulation of the stress-activated mitogen-activated protein kinase subfamilies in perfused heart. p38/RK mitogen-activated protein kinases and c-Jun N-terminal kinases are activated by ischemia/reperfusion. Circ Res 79:162–73
- Bulavin DV, Saito S, Hollander MC, et al. (1999). Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J 18:6845–54
- Cesura AM, Pinard E, Schubenel A, et al. (2003). The voltage-dependent anion channel is the target for a new class of inhibitors of the mitochondrial permeability transition pore. J Biol Chem 278:49812–18
- Clark JR, Sarafraz N, Marber MS. (2007). Potential of p38-MAPK inhibitors in the treatment of ischaemic heart disease. Pharmacol Ther 116:192–206
- Di Lisa F, Canton M, Memabo R, et al. (2007). Mitochondria and cardioprotection. Heart Fail Rev 12:249–60
- Gottlieb RA. (2011). Cell death pathways in acute ischemia/reperfusion injury. J Cardiovasc Pharmacol Ther 16:233–8
- Gottlieb RA, Burleson KO, Kloner RA, et al. (1994). Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest 94:1621–8
- Green DR, Reed JC. (1998). Mitochondria and apoptosis. Science 281:1309–12
- Halrestrap AP, Clarke SJ, Javadov SA. (2004). Mitochondrial permeability transition pore opening during myocardial reperfusion – A target for cardioprotection. Cardiovasc Res 61:372–85
- Hsu MJ, Hsu CY, Chem BC, et al. (2007). Apoptosis signal-regulating kinase 1 in amyloid beta peptide-induced cerebral endothelial cell apoptosis. J Neurosci 27:5719–29
- Jennings RB, Ganote CE. (1976). Mitochondrial structure and function in acute myocardial ischemic injury. Circ Res 38:I80–91
- Kim YA, Kim MY, Yu HY, et al. (2013). Gadd45beta is transcriptionally activated by p53 via p38alpha-mediated phosphorylation during myocardial ischemic injury. J Mol Med (Berl) 91:1303–13
- Kimura H, Shintani-ishida K, Nakajima M, et al. (2006). Ischemic preconditioning or p38 MAP kinase inhibition attenuates myocardial TNF alpha production and mitochondria damage in brief myocardial ischemia. Life Sci 78:1901–10
- Krijnen PA, Nijmeijer R, Meijer CJ, et al. (2002). Apoptosis in myocardial ischaemia and infarction. J Clin Pathol 55:801–11
- Kumphune S, Bassi R, Jacquet S, et al. (2010). A chemical genetic approach reveals that p38alpha MAPK activation by diphosphorylation aggravates myocardial infarction and is prevented by the direct binding of SB203580. J Biol Chem 285:2968–75
- Kumphune S, Chattipakorn S, Chattipakorn N. (2012). Role of p38 inhibition in cardiac ischemia/reperfusion injury. Eur J Clin Pharmacol 68:513–24
- Larsen JK, Yamboliev IA, Weber LA, Gerthoffer WT. (1997). Phosphorylation of the 27-kDa heat shock protein via p38 MAP kinase and MAPKAP kinase in smooth muscle. Am J Physiol 273:L930–40
- Li L, Tao JJ, Davaille J, et al. (2001). 15-Deoxy-delta 12,14-prostaglandin J2 induces apoptosis of human hepatic myofibroblasts. A pathway involving oxidative stress independently of peroxisome-proliferator-activated receptors. J Biol Chem 276:38152–8
- Lochner A, Marais E, Genade S, et al. (2009). Protection of the ischaemic heart: Investigations into the phenomenon of ischaemic preconditioning. Cardiovasc J Afr 20:43–51
- Lundberg KC, Szweda LI. (2004). Initiation of mitochondrial-mediated apoptosis during cardiac reperfusion. Arch Biochem Biophys 432:50–7
- Lv X, Wan J, Yang J, et al. (2008). Cytochrome P450 omega-hydroxylase inhibition reduces cardiomyocyte apoptosis via activation of ERK1/2 signaling in rat myocardial ischemia–reperfusion. Eur J Pharmacol 596:118–26
- Mcenery MW, Snowman AM, Trifiletti RR, Snyder SH. (1992). Isolation of the mitochondrial benzodiazepine receptor: Association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc Natl Acad Sci USA 89:3170–4
- Murphy KM, Ranganathan V, Farnsworth ML, et al. (2000). Bcl-2 inhibits Bax translocation from cytosol to mitochondria during drug-induced apoptosis of human tumor cells. Cell Death Differ 7:102–11
- O'Rouke B. (2000). Pathophysiological and protective roles of mitochondrial ion channels. J Physiol 529:23–36
- Perfettini JL, Castedo M, Nardacci R, et al. (2005). Essential role of p53 phosphorylation by p38 MAPK in apoptosis induction by the HIV-1 envelope. J Exp Med 201:279–89
- Premkumar A, Simantov R. (2002). Mitochondrial voltage-dependent anion channel is involved in dopamine-induced apoptosis. J Neurochem 82:345–52
- Saikumar P, Dong Z, Weinberg JM, Vemkatachalam MA. (1998). Mechanisms of cell death in hypoxia/reoxygenation injury. Oncogene 17:3341–9
- Schwertz H, Carter JM, Abdudureheman M, et al. (2007). Myocardial ischemia/reperfusion causes VDAC phosphorylation which is reduced by cardioprotection with a p38 MAP kinase inhibitor. Proteomics 7:4579–88
- Singhal PC, Bhaskaran M, Patel J, et al. (2002). Role of p38 mitogen-activated protein kinase phosphorylation and Fas-Fas ligand interaction in morphine-induced macrophage apoptosis. J Immunol 168:4025–33
- Son KN, Pugazhenthi S, Lipton HL. (2009). Activation of tumor suppressor protein p53 is required for Theiler's murine encephalomyelitis virus-induced apoptosis in M1-D macrophages. J Virol 83:10770–7
- Surinkaew S, Kumphune S, Chattipakorn S, Chattipakorn N. (2013). Inhibition of p38 MAPK during ischemia, but not reperfusion, effectively attenuates fatal arrhythmia in ischemia/reperfusion heart. J Cardiovasc Pharmacol 61:133–41
- Tanno M, Bassi R, Gorog DA, et al. (2003). Diverse mechanisms of myocardial p38 mitogen-activated protein kinase activation: Evidence for MKK-independent activation by a TAB1-associated mechanism contributing to injury during myocardial ischemia. Circ Res 93:254–61
- Thornton TM, Rincon M. (2009). Non-classical p38 map kinase functions: Cell cycle checkpoints and survival. Int J Biol Sci 5:44–51
- Thummasorn S, Kumfu S, Chattipakorn S, Chattipakorn N. (2011). Granulocyte-colony stimulating factor attenuates mitochondrial dysfunction induced by oxidative stress in cardiac mitochondria. Mitochondrion 11:457–66
- Vaseva AV, Marchenko ND, Ji K, et al. (2012). p53 Opens the mitochondrial permeability transition pore to trigger necrosis. Cell 149:1536–48
- Ward KW, Prokscht JW, Azzaranot LM, et al. (2001). Preclinical pharmacokinetics of SB-203580, a potent inhibitor of p38 mitogen-activated protein kinase. Xenobiotica 31:783–97
- World Health Organization. (2008). World Heath Statistics 2008 [Online]. Available from: http://www.who.int/entity/whosis/whostat/EN_WHS08_Full.pdf [last accessed 19 Apr 2014]