
Abstract
Context: Notoginsenoside R1 (NGR1) is the main component with cardiovascular activity in Panax notoginseng (Burk.) F. H. Chen, an herbal medicine that is widely used to enhance blood circulation and dissipate blood stasis.
Objective: The objective of this study is to investigate NGR1's effects on CYP1A2, CYP2C11, CYP2D1, and CYP3A1/2 activities in rats in vivo through the use of the Cytochrome P450 (CYP450) probe drugs.
Materials and methods: After pretreatment with NGR1 or physiological saline, the rats were administered intraperitoneally with a mixture solution of cocktail probe drugs containing caffeine (10 mg/kg), tolbutamide (15 mg/kg), metoprolol (20 mg/kg), and dapsone (10 mg/kg). The bloods were then collected at a set of time-points for the ultra-performance liquid chromatography/tandem mass spectrometry (UPLC-MS/MS) analysis.
Results: NGR1 was shown to exhibit an inhibitory effect on CYP1A2 by increased caffeine Cmax (43.13%, p < 0.01) and AUC0 − ∞ (40.57%, p < 0.01), and decreased CL/F (62.16%, p < 0.01) in the NGR1-treated group compared with those of the control group, but no significant changes in pharmacokinetic parameters of tolbutamide, metoprolol, and dapsone were observed between the two groups, indicating that NGR1 had no effects on rat CYP2C11, CYP2D1, and CYP3A1/2.
Discussion and conclusion: When NGR1 is co-administered with drugs that are metabolized by CYP1A2, the pertinent potential herb–drug interactions should be monitored.
Introduction
Cytochrome P450 (CYP450) is a superfamily of mixed-function oxygenases found in the liver and other tissues such as the gastrointestinal tract, which plays an important role in the metabolism of xenobiotics including drugs and chemicals, and endogenous compounds (Coon, Citation2005; Netsch et al., Citation2006). The expression of individual CYP450 is regulated by both endogenous factors and exogenous compounds including drugs and natural compounds. Herb–drug interactions can frequently occur when herb and a given chemical drug are co-administered, and the herbal medicine (one or more components) modulates the metabolism of the chemical drug by induction or inhibition of specific CYP450 isoform involved in its metabolism (Chavez et al., Citation2006; De Smet, Citation2007; Izzo, Citation2001). Although studies have investigated the effects of conventional drugs on CYP450 enzymes, only a small fraction of the thousands of medicinal plants used worldwide have been tested rigorously. Thus, very little is known about metabolic interactions between herbal medicines and conventional drugs.
Panax notoginseng (Burk.) F. H. Chen, a traditional herbal medicine that has been widely used in oriental countries for thousands of years, is now a popular natural medicine, either on its own or when mixed with other herbs in the treatment of microcirculatory disturbance-related diseases such as cardiovascular diseases, cerebral vascular diseases, and liver dysfunction (Jia et al., Citation2009; Liu et al., Citation2008; Park et al., Citation2005). Panax notoginseng saponins (PNS), as the major active component, can reduce inflammatory reactions in patients and animal models (Feng et al., Citation2004; Ning et al., Citation2012), and can also ameliorate the symptoms of disseminated intravascular coagulation (Wang, Citation2012; Yang et al., Citation2005; Zhang et al., Citation2012). Notoginsenoside R1 (NGR1, ) is considered as one of the main active ingredients in PNS and has been widely applied to treat angina pectoris and myocardial infarction. However, little is known about its effects on CYP450 and potential interactions are not predictable when it is co-administered with other drugs (Chen et al., Citation2002; Zhang & Wang, Citation2006). The purpose of the study was to determine the possible effects of NGR1 on the main four CYP450 subtypes (CYP1A2, CYP2C11, CYP2D1, and CYP3A1/2) in rats, using caffeine (CYP1A2), tolbutamide (CYP2C11), metoprolol (CYP2D1), and dapsone (CYP3A1/2) as the probe substrates. The results should help to predict possible herb–drug interactions mediated by CYP450 and decrease the incidence of the interactions.
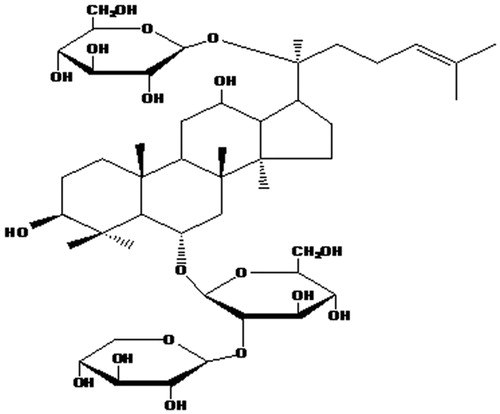
Figure 1. Chemical structure of NGR1.
Materials and methods
Materials and reagents
NGR1 was obtained from Chengdu Mansite Pharmaceutical Co. (Chengdu, China). Caffeine, metoprolol, and phenacetin were purchased from National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Dapsone was purchased from Sigma Chemical Co. (St Louis, MO). Tolbutamide was obtained from Dr. Ehrenstorfer (Augsburg, Germany). Acetonitrile (LC-grade), methanol, and formic acid (HPLC-grade) were purchased from the Sigma Aldrich Co. (St Louis, MO). Dichloromethane and n-butanol (analytical grade) were supplied by Tianjin Fuyu Fine Chemicals Co., Ltd. (Tianjin, China). Polysorbate 80 was obtained from Changsha Chemical Reagent Co. (Changsha, China). All the other reagents were of analytical grade. The mixture solution of probe drugs for administration was prepared in physiological saline that contained 2% polysorbate 80 to dissolve all drugs completely.
Animals and treatment
All animal study protocols were pre-approved by the Experimental Animal Ethics Committee of Harbin Medical University, China (Animal Experimental Ethical Inspection Protocol no. HMUIRB20120011). Male Wistar rats (body weight 180 ± 20 g) were supplied by the Animal Experimental Center of the Harbin Medical University (Harbin, China), which is an accredited institution authenticated by the Institutional Animal Care and Use Committee. The rats were handled in a manner that met all the recommendations of the National Society for Medical Research and Guidelines for the Care and Use of Laboratory Animals. Totally 16 rats were divided randomly into two groups of eight, the blank control group and the NGR1-treated group. The two groups were administrated physiological saline and 10 mg/kg NGR1 (2 mL) that was prepared in physiological saline by caudal vein for seven consecutive days. On day 8, the mixture solution of probe drugs was administrated to the animals in both groups with caffeine (10 mg/kg), tolbutamide (15 mg/kg), metoprolol (20 mg/kg), and dapsone (10 mg/kg) by intraperitoneal injection. Blood samples (300 μL) were collected by caudal vein at 0, 0.17, 0.5, 0.83, 1.17, 1.5, 2, 3, 5, 8, 12, and 24 h after administration of the drugs and were put into heparinized test tubes. After centrifugation at 4000 × g for 10 min, the plasma samples were obtained and preserved at −20 °C for further analysis.
Sample treatment
Phenacetin standard (internal standard) solution (100 μg/mL, 20 μL) and 2 mL extraction mixture (dichloromethane: n-butanol, 10:1, v/v) were added to fresh blood plasma (100 μL). Each sample was mixed thoroughly by vortex for 2 min and centrifuged at 3000 × g for 10 min. Then, the organic phase of 1.8 mL was taken out and evaporated to dryness under nitrogen at 40 °C in water bath. The residue was dissolved in 200 μL mobile phase and an aliquot of 10 μL was injected into the UPLC-MS/MS system for analysis.
UPLC-MS/MS measurement
The chromatographic separation was carried out using a Waters Acquity (Waters, Milford, MA) UPLC and performed on a Waters Acquity UPLC BEH HILIC C18 column (1.7 μm, 2.1 × 50 mm; Waters, Milford, MA). The column temperature was maintained at 40 °C. The sampler chamber temperature in the auto sampler was kept at 10 °C. The mobile phase consisted of an acetonitrile with 0.1% formic acid (15:85, v/v) at a flow rate of 0.25 mL/min. The total run time for each injection was 6 min. Waters MS (Waters MS Technologies, Manchester, UK) and a micromass Quattro micro API triple quadrupole tandem mass spectrometer were equipped with an electrospray ionization (ESI) source. The ESI source was set to a positive ion mode with ionization conditions as follows: capillary voltage 3.3 kV, cone voltage 30 V, source temperature 120 °C, and desolvation gas (nitrogen) temperature 350 °C (650 L/h). Quantification was performed using multiple reaction monitoring (MRM) transitions of m/z 249 → 92 (dapsone), m/z 195 → 138 (caffeine), m/z 268 → 116 (metoprolol), m/z 271 → 91 (tolbutamide), and m/z 180 → 110 (phenacetin, internal standard). Instrument control and data acquisition were performed with MassLynxTM V 4.1 software (Waters, Milford, MA). The regression equations and the lower limit of quantitation (LLOQ) concentrations of the analytes are shown in .
Table 1. Regression equation, linear range, low limits of quantification (LLOQ) for probe substrates in rat plasma.
Statistical analysis
Data were expressed as means ± SD. Pharmacokinetic parameters of the four probe drugs were derived with a non-linear regression iterative program, DAS 2.0 (Chinese Pharmacological Society, Beijing, China) pharmacokinetic statistical software. The peak plasma concentration (Cmax) and the time to reach the peak plasma concentration (tmax) were obtained directly from the concentration–time data. The elimination rate constant (Ke) was calculated from the slope of the logarithm of the plasma concentration versus time date using the final four points. The apparent elimination half-time (t1/2) was calculated as 0.693/Ke. The area-under-the-concentration–time curve (AUC) was calculated according to the non-compartmental trapezoidal method, with extrapolation to infinity from the terminal slope of the fitted curve. The total plasma clearance (CL/F) was calculated as Dose/AUC.
Statistically significant differences in the pharmacokinetic parameters between the NGR1-treated group and the blank control group were assessed by ANOVA and Dunnett's test, p < 0.05 and p < 0.01 were considered to be statistically significant and highly significant, respectively.
Results
Effect of NGR1 on CYP1A2
The blood concentration–time curves and pharmacokinetics parameters of caffeine are shown in and , respectively. According to the results, NGR1 inhibited the metabolism of caffeine as suggested, by increased Cmax (43.13%, p < 0.01) and AUC0–∞ (40.57%, p < 0.01), and decreased CL/F (62.16%, p < 0.01) in the NGR1-treated group as compared with those of the blank control group.
Figure 2. Mean plasma concentration–time profiles of caffeine in rats after intraperitoneal administration of 10 mg/kg caffeine in the blank control group and the NGR1-treated group.
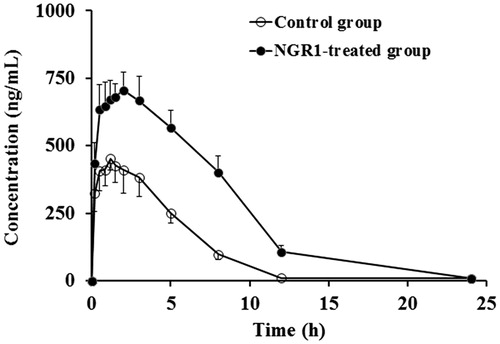
Table 2. Pharmacokinetic parameters of caffeine, tolbutamide, metoprolol, and dapsone after intraperitoneal administration to rats pretreated with physiological saline or NGR1.
Effect of NGR1 on CYP2C11
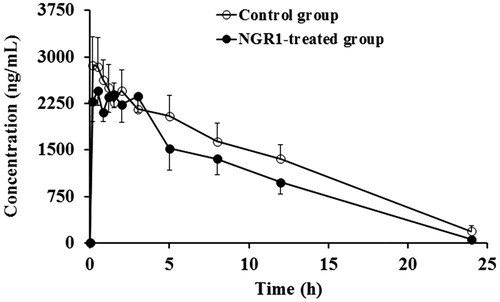
The data in and showed that CYP2C11 was not affected by NGR1, for Cmax, AUC and tmax of tolbutamide were not significantly altered in the NGR1-treated group as compared with those of the blank control group.
Figure 3. Mean plasma concentration–time profiles of tolbutamide in rats after intraperitoneal administration of 15 mg/kg tolbutamide in the blank control group and the NGR1-treated group.
Effect of NGR1 on CYP2D1
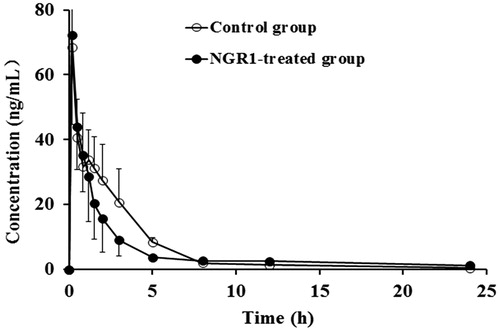
According to the concentration–time curves and pharmacokinetics parameters ( and ), there were no significant differences in the major pharmacokinetics parameters of metoprolol between the NGR1-treated group and the blank control group.
Figure 4. Mean plasma concentration–time profiles of metoprolol in rats after intraperitoneal administration of 20 mg/kg metoprolol in the blank control group and the NGR1-treated group.
Effect of NGR1 on CYP3A1/2
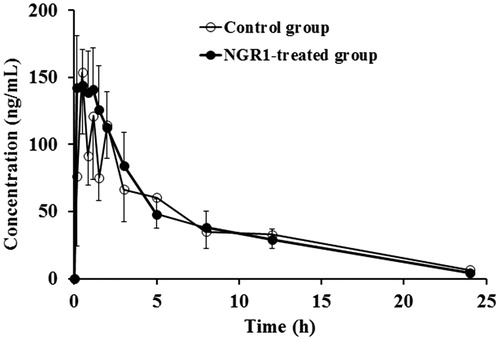
The mean dapsone plasma concentration–time profiles were overlaid by the blank control group and the NGR1-treated group (). The pharmacokinetics parameters of dapsone are shown in . The data showed that there were no significant differences between the NGR1-treated group and the blank control group.
Figure 5. Mean plasma concentration–time profiles of dapsone in rats after intraperitoneal administration of 10 mg/kg dapsone in the blank control group and the NGR1-treated group.
Statistical results of pharmacokinetics parameters
The statistical results of the pharmacokinetics parameters of the NGR1-treated group and the blank control group are shown in . The data showed that all the pharmacokinetics parameters (Cmax, t1/2, CL/F, AUC0 − t, and AUC0 − ∞) of caffeine were varied significantly (p < 0.01) in the NGR1-treated group compared with those of the blank control group. No significant differences were found for the pharmacokinetic parameters of tolbutamide, metoprolol, and dapsone.
Table 3. p value of pharmacokinetics parameters.
Discussion
The study demonstrated the inhibitory effect of NGR1 on CYP1A2 in rats. After pretreatment with NGR1, the plasma concentrations of caffeine increased significantly and the metabolism of caffeine in the NGR1-treated group was slower than that of the blank control group. Caffeine is the probe substrate of CYP1A2 which is involved in the metabolism of many drugs. CYP1A2 amounts for 13% of the total CYP450 content and plays an important role in the metabolic clearance of approximately 5% of currently marketed drugs, such as paracetamol, diazepam, lidocaine, clarithromycin, diethylstilbestrol, verapamil, nifedipine, propranolol, warfarin, and theophylline (Arold et al., Citation2005; Diani-Moore et al., Citation2006; Fukami et al., Citation2007; Khanm & Preskorn, Citation2005; Ou-Yang et al., Citation2000; Xiao & Zhou, Citation2008; Zhang et al., Citation1995). Studies have reported the effects on CYP1A2 by some herbs (Hu et al., Citation2010; Ueng et al., Citation2010; von Moltke et al., Citation2004). The results of the present study could speculate that inhibition of CYP1A2 by NGR1 may result in elevated plasma concentration of co-administered drugs that are substrates of CYP1A2, which may cause adverse side effects due to the concentration of concomitantly used drugs over the limit of the toxicity threshold, especially for the drugs with narrow therapeutic windows.
The study also focused on the effects of NGR1 on the activities of other three major CYP450 isoforms containing CYP2C11, CYP2D1, and CYP3A1/2. Together with CYP1A2, these isoforms are involved in the metabolism of an estimated 90% of prescription and non-prescription medications (Moorthy, Citation2008; van Schaik & Ron, Citation2008). Tolbutamide, metoprolol, and daspone are the probe substrates for CYP2C11, CYP2D1, and CYP3A1/2, respectively (Sharma et al., Citation2004; Turpault et al., Citation2009). Our results showed that CYP2C11, CYP2D1, and CYP3A1/2 activities were not significantly affected by NGR1, no significant differences were observed in the AUC, CL/F, t1/2, tmax, or Cmax of tolbutamide, metoprolol, and dapsone between the NGR1-treated group and the blank control group. Therefore, it is speculated that NGR1 is unlikely to have clinically relevant effects on the disposition of drugs primarily dependent on CYP2C11, CYP2D1, or CYP3A1/2 pathways for metabolism.
It is reported that PNS has a wide variety of therapeutic uses when it comes to promote blood circulation, remove blood stasis, inhibit platelet aggregation, and protect against damage that results from myocardial ischemia (Ng et al., Citation2006), resist arrhythmia, relieve swelling, and alleviate pain (Chan et al., Citation2002b; Cicero et al., Citation2003; Zhang & Jin, Citation2006). It has also been used for the treatment of coronary heart disease and cerebral vascular disease such as angina pectoris and myocardial infarction with favorable results (Chan et al., Citation2002a). As the major active ingredient of PNS, NGR1 performs antioxidant, anti-inflammatory, antiapoptotic, and immune stimulatory activities (Deng et al., Citation2009; He et al., Citation2012).
As a typical and traditional herb medicine, PNS has been made into preparation and widely used in clinic. The effects of PNS preparation on CYP450 enzymes have been studied. Liu reported that Xueshuantong injection (PNS injection) showed no effect on CYP3A4 and CYP1A2 (Liu et al., Citation2010). The effect on CYP3A4 in our study of NGR1 is the same as the Xueshuantong injection. However, for the effect on CYP1A2, our result is not the same. The reason may be that PNS includes more than 30 different saponins (Ruan et al., Citation2010; Sun et al., Citation2007), including Ginsenoside Rb1, Rg1, and NGR1 as its main active compounds (Wang et al., Citation2007). These saponins may have different effects on CYP450 enzymes, and the effect of PNS injection on CYP450 enzymes is a comprehensive result of all the saponins.
Some studies have reported effects of certain compounds of PNS on CYP450 enzymes up to now. For example, Henderson et al. (Citation1999) reported the effects of ginsenoside Rb1 and Rg1 on the CYP450 isoforms and suggested that ginsenoside Rb1 and Rg1 had no effects on CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. However, no study has evaluated the impact of NGR1 on the activities of CYP450 isoforms. For the first time, our present study investigated the effects of NGR1 on the activities of the four main CYP450 isoforms CYP1A2, CYP2C11, CYP2D1, and CYP3A1/2, and demonstrated the inhibitory effect of NGR1 on CYP1A2, but no effect on the activities of CYP2C11, CYP2D1, and CYP3A1/2 in rats in vivo.
In clinical situations, herbal medicine and chemical drugs may be administered concomitantly. The metabolic drug interactions may occur when a herbal medicine and a given chemical drug are co-administered if the herbal medicine modulates the metabolism of the chemical drug by induction or inhibition on CYP450 isoforms (Foster et al., Citation2002; Gao et al., Citation2012). These drug interactions have become more and more common as a result of polypharmacy and the increasing use of alternative medicine, including herbs and natural products (Dou et al., Citation2011; Venkataramanan et al., Citation2006; Zhou et al., Citation2003). Due to the increasing use of herbal medicines, often in combination with conventional therapeutic drugs, it is important to identify the effects of herbal preparations or natural products on CYP450 activity and whether these products have the potential to affect the pharmacokinetics of drugs which rely on CYP450 for their metabolism and clearance. Since the activity of CYP1A2 has been inhibited by NGR1, it is necessary to study the interactions between drugs metabolized by CYP1A2 and NGR1 preparations.
Conclusion
Our study showed that NGR1 inhibited the activity of CYP1A2, but had no effects on activities of CYP2C11, CYP2D1, and CYP3A1/2. CYP1A2 is responsible for the metabolism and disposition of a large number of drugs currently used, the potential herb–drug interactions of NGR1 preparations with drugs that serve as substrates of CYP1A2 may be important. Further investigation for the clinical significance is needed.
Declaration of interest
The authors report that they have no conflicts of interest. The authors alone are responsible for the content and writing of this article. This work was supported by Natural Science Foundation of Heilongjiang [D201044] and National Natural Science Foundation of China [81173659].
References
- Arold G, Donath F, Maurer A, et al. (2005). No relevant interaction with alprazolam, caffeine, tolbutamide, and digoxin by treatment with a low-hyperforin St John's Wort extract. Planta Med 71:331–7
- Chan P, Thomas GN, Tomlinson B. (2002a). Protective effects of trilinolein extracted from Panax notoginseng against cardiovascular disease. Acta Pharmacol Sin 23:1157–62
- Chan RY, Chen WF, Dong A, et al. (2002b). Estrogen-like activity of ginsenoside Rg1 derived from Panax notoginseng. J Clin Endocrinol Metab 87:3691–5
- Chavez ML, Jordan MA, Chavez PI. (2006). Evidence-based drug–herbal interactions. Life Sci 78:2146–57
- Chen CH, Li XL, Zhang JX, et al. (2002). The effects of R1 and Rd extracted from Panax notoginseng saponins on microcirculation and blood coagulation. J West China Univ Med Sci 33:550–2
- Cicero AF, Vitale G, Savino G, et al. (2003). Panax notoginseng (Burk.) effects on fibrinogen and lipid plasma level in rats fed on a high-fat diet. Phytother Res 17:174–8
- Coon MJ. (2005). Cytochrome P450: Nature's most versatile biological catalyst. Annu Rev Pharmacol Toxicol 45:1–25
- De Smet PA. (2007). Clinical risk management of herb–drug interactions. Br J Clin Pharmacol 63:258–67
- Deng GF, Wang DL, Meng MX, et al. (2009). Simultaneous determination of notoginsenoside R1, ginsenoside Rg1, Re, Rb1 and icariin in rat plasma by ultra-performance liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 877:2113–22
- Diani-Moore S, Papachristou F, Labitzke E, et al. (2006). Induction of CYP1A and cyp2-mediated arachidonic acid epoxygenation and suppression of 20-hydroxyeicosatetraenoic acid by imidazole derivatives including the aromatase inhibitor vorozole. Drug Metab Dispos 34:1376–85
- Dou DQ, Xiang Z, Yang G, et al. (2011). Prediagnostic methods for the hemolysis of herbal medicine injection. J Ethnopharmacol 138:445–50
- Feng L, Xiong Y, Cheng F, et al. (2004). Effect of ligustrazine on ischemia–reperfusion injury in murine kidney. Transplant Proc 36:1949–51
- Foster BC, Vandenhoek S, Tang R, et al. (2002). Effect of several Chinese natural health products of human cytochrome P450 metabolism. J Pharm Pharm Sci 5:185–9
- Fukami T, Nakajima M, Sakai H, et al. (2007). CYP2A13 metabolizes the substrates of human CYP1A2, phenacetin and theophylline. Drug Metab Dispos 35:335–9
- Gao LC, Zhang W, Liu ZQ, et al. (2012). Progress in clinical herb–drug interactions mediated by drug metabolism-related genes. Chin J Clin Pharmacol Ther 3:346–51
- He NW, Zhao Y, Guo L, et al. (2012). Antioxidant, antiproliferative, and pro-apoptotic activities of a saponin extract derived from the roots of Panax notoginseng (Burk.) F.H. Chen. J Med Food 15:350–9
- Henderson GL, Harkey MR, Gershwin ME, et al. (1999). Effects of ginseng components on cDNA-expressed cytochrome P450 enzyme catalytic activity. Life Sci 65:209–14
- Hu L, Xu W, Zhang X, et al. (2010). In-vitro and in-vivo evaluations of cytochrome P450 1A2 interactions with nuciferine. J Pharm Pharmacol 62:658–62
- Izzo AA, Ernst E. (2001). Interactions between herbal medicines and prescribed drugs. Drugs 15:2163–75
- Jia L, Zhao Y, Liang XJ. (2009). Current evaluation of the millennium phytomedicine-ginseng (II): Collected chemical entities, modern pharmacology, and clinical applications emanated from traditional Chinese medicine. Curr Med Chem 16:2924–42
- Khanm AY, Preskorn SH. (2005). Examining concentration-dependent toxicity of clozapine: Role of therapeutic drug monitoring. J Psychiatr Pract 11:289–301
- Liu L, Shi R, Shi Q, et al. (2008). Protective effect of saponins from Panax notoginseng against doxorubicin-induced cardiotoxicity in mice. Planta Med 74:203–9
- Liu SJ, Ju WZ, Chen WK, et al. (2010). Influence of xue-shuan-tong injection on P450 activities using a cocktail method. J Chin Pharm 45:115–18
- Moorthy B. (2008). The CYP1A Subfamily. In: Ioannides C, ed. Cytochromes P450 Role in the Metabolism and Toxicity of Drugs and other Xenobiotics. Cambridge: Royal Society of Chemistry, 101
- Netsch MI, Gutmann H, Schmidlin CB, et al. (2006). Induction of CYP1A by green tea extract in human intestinal cell lines. Planta Med 72:514–20
- Ng TB. (2006). Pharmacological activity of sanchi ginseng (Panax notoginseng). J Pharm Pharmacol 58:1007–19
- Ning N, Dang X, Bai C, et al. (2012). Panax notoginsenoside produces neuroprotective effects in rat model of acute spinal cord ischemia-reperfusion injury. J Ethnopharmacol 139:504–12
- Ou-Yang DS, Huang SL, Wang W, et al. (2000). Phenotypic polymorphism and gender-related differences of CYP1A2 activity in a Chinese population. Br J Clin Pharmacol 49:145–51
- Park WH, Lee SK, Kim CH. (2005). A Korean herbal medicine, Panax notoginseng, prevents liver fibrosis and hepatic microvascular dysfunction in rats. Life Sci 76:1675–90
- Ruan JQ, Leong WI, Yan R, et al. (2010). Characterization of metabolism and in vitro permeability study of notoginsenoside R1 from radix notoginseng. J Agric Food Chem 58:5570–6
- Sharma A, Pilote S, Belanger PM, et al. (2004). A convenient five-drug cocktail for the assessment of major drug metabolizing enzymes: A pilot study. Br J Clin Pharmacol 58:288–97
- Sun K, Wang CS, Guo J, et al. (2007). Protective effects of ginsenoside Rb1, ginsenoside Rg1, and notoginsenoside R1 on lipopolysaccharide-induced microcirculatory disturbance in rat mesentery. Life Sci 8:509–18
- Turpault S, Brian W, Van Horn R, et al. (2009). Pharmacokinetic assessment of a five-probe cocktail for CYPs 1A2, 2C9, 2C19, 2D6 and 3A. Br J Clin Pharmacol 68:928–35
- Ueng YF, Tsai CC, Lo WS, et al. (2010). Induction of hepatic cytochrome P450s by the herbal medicine Sophora flavescens extract in rats: Impact on the elimination of theophylline. Drug Metab Pharmacokinet 25:560–7
- van Schaik, Ron HN. (2008). CYP450 pharmacogenetics for personalizing cancer therapy. Drug Resist Update 3:77–98
- Venkataramanan R, Komoroski B, Strom S. (2006). In vitro and in vivo assessment of herb drug interactions. Life Sci 78:2105–15
- von Moltke LL, Weemhoff JL, Bedir E, et al. (2004). Inhibition of human cytochromes P450 by components of Ginkgo biloba. J Pharm Pharmacol 56:1039–44
- Wang JB, Li SP, Chen JM, et al. (2007). Chemical characteristics of three medicinal plants of the Panax genus determined by HPLC-ELSD. J Sep Sci 30:825–32
- Wang Y. (2012). Pharmacological effects of sanqi in blood system and new clinical application. Clin J Chin Med 4:45
- Xiao P, Zhou HH. (2008). Advance in cytochrome P450 1A2. J Cent South Univ (Med Sci) 5:456–60
- Yang ZG, Chen AQ, Yu SD, et al. (2005). Recent advances in the research of pharmacological activities of Panax notoginseng saponins. Chin J Veteran Drug 39:33–7
- Zhang HS, Wang SQ. (2006). Notoginsenoside R1-from Panax notoginseng inhibits TNF-alpha-induced PAI-1 production in human aortic smooth muscle cells. Vascul Pharmacol 44:224–30
- Zhang HY, Jin JS. (2006). Progress of Panax notoginseng in pharmacological and clinical research for nephropathy. Med Recapitul 12:1466–7
- Zhang W, Chen G, Deng CQ. (2012). Effects and mechanisms of total Panax notoginseng saponins on proliferation of vascular smooth muscle cells with plasma pharmacology method. J Pharm Pharmacol 64:139–44
- Zhang Z, Fasco MJ, Huang Z, et al. (1995). Human cytochromes P4501A1 and P4501A2: R-warfarin metabolism as a probe. Drug Metab Dispos 23:1339–46
- Zhou S, Gao Y, Jiang W, et al. (2003). Interactions of herbs with cytochrome P450. Drug Meta Rev 35:35–98