
Abstract
Context: Nicotine is an abundant and most significant component of cigarette smoke. Epidemiological evidence strongly suggests an association between cigarette smoking and pancreatic injury, although effects of smoking on endocrine pancreas are still controversial.
Objective: We examined the impact and underlying mechanisms of action of folic acid and vitamin B12 on nicotine-induced damage in pancreatic islets of rats.
Materials and methods: Male Wistar rats were treated with nicotine (3 mg/kg body weight/d, intraperitonealy) with or without folic acid (36 µg/kg body weight/d, orally) and vitamin B12 (0.63 µg/kg body weight/d, orally) for 21 d. Fasting blood glucose, oral glucose tolerance test, HBA1c, insulin, oxidative stress parameters, proinflammatory cytokines, and CRP level were measured. Histological evaluation, TUNEL assay, and immunohistochemical staining of NF-κB and caspase-3 were also performed.
Results: Folic acid and vitamin B12 blunted the nicotine-induced impairment in fasting blood glucose (51–56% recovery), HbA1c (64–76% recovery), oral glucose tolerance, insulin level (23–40% recovery), and islet cell counts (26–74% recovery) in rats. Moreover, folic acid in combination with vitamin B12 also attenuated the nicotine-induced changes in markers of oxidative stress (17–88% recovery), TNF-α (40–99% recovery), and IL-6 level (47–65% recovery), CRP level (59–73% recovery), expression of NF-κB and caspase-3, and apoptosis in pancreatic islet cells.
Discussion and conclusion: The present study shows that folic acid and vitamin B12 supplementation can reduce nicotine-induced impairment in glucose homeostasis and apoptosis and damage of pancreatic islet cells by modulating oxidative stress, levels of proinflammatory cytokines, and expression of NF-κB.
Introduction
Nicotine, a major component of tobacco and cigarette smoke, is an addictive agent and has been characterized as a drug of abuse by the US Surgeon General (Momi et al., Citation2012). Approximately, 430 000 people die of causes related to smoking cigarettes and nearly 30% of those deaths are due to some form of cancer (Okuyemi, Citation2001). The economic burden due to abuse of this drug is substantial because of the well-documented pathophysiological effects of nicotine on organs in the cardiovascular, respiratory, hepatic, renal, and nervous systems.
Diabetes mellitus and lesser forms of glucose intolerance, particularly impaired glucose tolerance, can now be found in almost every population in the world and epidemiological evidence suggests that, without effective prevention and control programs, diabetes will likely continue to increase globally. On the other hand, by the year 2020, tobacco will become the single leading cause of death, causing one out of every eight deaths (Gupta, Citation2001). The alkaloid nicotine is currently available in various formulations, such as chewing gum, topical patches, and nasal spray to stop the smoking habit (Balfour et al., Citation2000; Valenca et al., Citation2008). So, a combination of nicotine and diabetes poses a serious threat to human mankind. There are several reports suggesting that smoking may increase the risk of type 2 diabetes (Radzeviciene & Ostrauskas, Citation2009; Yeh et al., Citation2010). The exact mechanism behind the association between smoking and diabetes and its complications is not clear. Although smoking can stimulate the release of counter-regulatory hormones and cause an elevation in plasma glucose levels, the effects of smoking on insulin secretion and insulin resistance are, however, controversial. There are reports suggesting a decrease in fasting insulin level in smokers while other reports showed an unimpaired or somewhat over stimulated insulin secretion in smokers (Attvall et al., Citation1993; Facchini et al., Citation1992). These inconsistent clinical results are not well understood. The study by Daniel and Cargo (Citation2004) on aboriginal people who have a high prevalence of smoking and diabetes indicates that the toxic effect of smoking on pancreatic cells may be somehow variable in different conditions. In addition, there was an in vitro study with rat and human islets showing acute or 48 h exposure to nicotine might moderately inhibit the insulin release (Yoshikawa et al., Citation2005).
On the contrary, increasing evidence suggests an acute effect of smoking on impairing insulin action and hence induction of an insulin resistant state in both diabetic and non-diabetic subjects (Attvall et al., Citation1993). Although the effects are reported to be only acute and are reversible, repeated increases in insulin resistance and plasma glucose due to habitual smoking might increase smokers' vulnerability to develop frank diabetes (Daniel & Cargo, Citation2004).
Nicotine has been recognized to result in oxidative stress by inducing the generation of reactive oxygen species (ROS) (Mahapatra et al., Citation2010). These ROS in turn are capable of initiating and promoting oxidative damage in the form of lipid peroxidation; protein oxidation, and DNA damage (Das et al., Citation2009). Lipid peroxidation is known to cause cellular injury by inactivation of membrane enzymes and receptors, depolymerization of polysaccharide, as well as protein cross-linking and fragmentation (Luqman & Rizvi, Citation2006). The immune cells use ROS for carrying out their normal functions, but an excess amount of ROS can attack cellular components that lead to cell damage. Previous reports have shown that nicotine administration results in the imbalance of prooxidant/antioxidant status in different tissues of Wister rats (Muthukumaran et al., Citation2008; Neogy et al., Citation2008).
Pancreatic islet cells are highly susceptible to the toxic effects of reactive oxygen intermediates (ROIs) and NO, activation of poly (ADP-ribose) polymerase (PARP), and cellular NAD depletion to induce the death of several cell types, including the loss of insulin-producing islet cells in type I diabetes (Mukherjee et al., Citation2006). Furthermore, islets appear to be more vulnerable to oxidative injury than other tissues because they express low levels of antioxidant enzymes, including superoxide dismutase (SOD), which converts superoxide radicals to H2O2, and catalase (CAT) and cellular glutathione peroxidase (Gpx-1), which detoxify H2O2 (Mysore et al., Citation2005). Over expression of a combination of antioxidant enzymes in islets may provide the most effective protection from ROS-mediated injury (Lortz & Tiedge, Citation2003).
Biological compounds with antioxidant properties contribute to the protection to cells and tissues against deleterious effects of ROS and other free radicals (Liu et al., Citation2013). Folic acid is a vitamin of the B group and is converted in vivo through a series of enzymatic transformations into N5, N10-dimethylene-5,6,7,8-tetrahydrofolate, which is a coenzyme (Joshi et al., Citation2001). It has been shown that folic acid can protect bioconstituents from free radical damage at least by competition, which otherwise can lead to oxidative stress (Khan, Citation2002). In spite of being a water-soluble molecule, folic acid can inhibit lipid peroxidation also. The scavenging and repair of thiyl radicals by folic acid make it a potential vitamin to be called as an antioxidant (Khan, Citation2002). Cigarette smoking is known to be associated with raised homocysteine levels (Reis et al., Citation2000). Smokers also tend to have lower levels of folic acid and vitamin B12 (McCarty, Citation2000; Pagan et al., Citation2001), all of which affect homocysteine levels by acting as co-enzyme, i.e., vitamins B12 or co-substrate, i.e., folic acid for the enzymes controlling homocysteine metabolism (Muzawar & Patil, Citation2011). Several cross-sectional studies (Hermann et al., Citation2004; Nervo et al., Citation2011; Pflipsen et al., Citation2009) and case reports (Bell, Citation2010; Liu et al., Citation2006) have documented an increased frequency of vitamin B12 deficiency among type 2 DM (T2DM) patients. There are no guidelines to address how often patients with T1DM and T2DM should be supplemented with vitamin B12. The optimal supplementation dose of vitamin B12 is also unknown.
So the goal of this study was to investigate the extent of nicotine-induced oxidative stress-mediated islet cell damage in rats and, further, to examine whether the supplementation with folic acid and vitamin B12 may be an effective and viable strategy for reducing nicotine-induced oxidative stress and preventing pancreatic islet cell damaging effects on exposure to nicotine.
Materials and methods
Animal model
All animal experiments were performed according to the ethical guidelines suggested by the Institutional Animal Ethics Committee (IAEC). Male Wistar albino rats weighing 110–125 g were used in all the experiments. Animals were maintained in an environmentally controlled animal house (temperature 24 ± 3 °C) and in a 12 h light/dark schedule with free access to water supply.
Experimental design
For experiments, rats were randomly divided into four groups consisting of five rats in each: Group A, control; Group B, nicotine-treated; Group C, nicotine + folic acid; Group D, nicotine + folic acid + vitamin B12 supplemented. The animals of all groups were provided with a control diet composed of 71% carbohydrate, 18% protein, 7% fat, and 4% salt mixture (Mukherjee et al., Citation2006). The dose and the administration route of nicotine were used as reported earlier (Chattopadhyay & Chattopadhyay, Citation2008). Animals in Groups B, C, and D received intraperitoneal injection of nicotine tartrate (dissolved in 0.9% physiological saline) at a dose of 3.0 mg/kg body weight daily for 21 d at 16:00 h every day to avoid diurnal variation. The dilution was done in such a way that 1 ml of physiological saline contained the required dose of nicotine. Simultaneously, animals in the control subgroup received intraperitoneal injection of 1 ml physiological saline. In addition, animals of Groups C and D, respectively, were orally treated with folic acid (36 µg/kg body weight/d for 21 d) only or folic acid and vitamin B12 (0.63 µg/kg body weight/d for 21 d) simultaneously one after another (Mukherjee et al., Citation2006). To overcome the impact of any altered food intake, animals of Group A were pair-fed with experimental groups B, C, and D.
Determination of fasting blood glucose and OGTT
After the experimental period was over (21 d), the animals were kept in overnight fasting. In the next morning, blood samples were collected from retro-orbital veins. After collection of blood, the animals of both groups were orally administered with 1 g of glucose/kg body weight. Thereafter, blood samples were collected at every 30 min interval for 2 h and 30 min. Glucose oxidase enzyme kit (E. Merck, Mumbai, India) was used for the estimation of blood glucose from all samples.
Determination of HbA1C
Blood samples collected were mixed with EDTA (as per specification of the kit) and were used for estimation of glycated hemoglobin (HBA1c) following the ion-exchange resin-based method.
Serum and plasma preparation
After the collection of blood for fasting blood glucose, HBA1c, and OGTT, all the rats were anesthetized with pentobarbitone sodium (60 mg/kg body weight) and sacrificed by cervical dislocation. Blood samples were drawn from heart in two separate test tubes: one with anticoagulant and another without anticoagulant. Serum and plasma were separated for the C-reactive protein (CRP) and insulin assay, respectively.
Determination of insulin
Plasma insulin was measured by enzyme-linked imunosorbent assay (ELISA) using the kit (Cayman Chemicals, Ann Arbor, MI). The intra assay variation was 4.9%. As the samples were run at a time, there was no inter-assay variation. The level of insulin in plasma was expressed in μIU/ml.
Preparation of pancreatic tissue extract
The abdomen was opened, and the pancreas was quickly removed. For the estimation of NO, MDA, and SOD, pancreatic tissue extract was prepared in ice-cold Tris-Hcl buffer (pH 7.4) (Mukherjee et al., Citation2006). For CAT and GSH estimations, the tissues were homogenized in ice-cold isotonic phosphate buffer (pH 7.0 and pH 8.0, respectively).
Estimation of nitric oxide production and lipid peroxidation
The role of nitric oxide synthase (NOS) was indirectly assessed by estimating the amount of NO production. Nitric oxide decomposes rapidly in aerated solutions to form stable nitrite/nitrate products. Nitrite accumulation was estimated by Griess reaction (Raso et al., Citation1999) and was used as an index of NO production in this study. The amount of nitrite in the sample (micromolar unit) was calculated from a sodium nitrite standard curve.
The role of lipid peroxidase was assessed by studying the level of formation of malondialdehyde (MDA), an indicator of lipid peroxidation. Quantitative measurement of lipid peroxidation was performed following the thiobarbituric acid (TBA) test (Wills, Citation1987). The amount of MDA formed was quantitated with TBA and used as an index of lipid peroxidation. The results were expressed as nanomoles of MDA per milligram of protein using the molar extinction coefficient (1.56 × 105 cm2/mM).
Estimation of superoxide dismutase
The nitroblue tetrazolium (NBT) method based on the inhibition of NBT reduction by SOD was used for the determination of SOD activities (Sun et al., Citation1988). The relative absorbance was then converted into unit of SOD activity per mg protein, where one unit of SOD activity was equivalent to the quantity of SOD that caused a 50% reduction in the background rate of NBT reduction.
Estimation of catalase
Catalase activity was determined according to the method of Aebi (Citation1984) by following the decomposition of H2O2 at 240 nm. The difference in absorbance per unit time was used as a measure of CAT activity. The values were expressed as U/mg protein.
Estimation of glutathione level
GSH level was assayed using 5,5′-dithiobis-2-nitrobenzoic acid (DTNB). The absorbance of reduced chromogen was followed spectrophotometrically at 412 nm. GSH level was determined from a standard curve and expressed as mM/mg protein (Sedlak & Lindsay, Citation1968).
Estimation of tissue protein content
Tissue protein content was determined according to the method developed by Lowry et al. (Citation1961) using bovine serum albumin as a standard.
Preparation of permanent slides for histological and immunocytochemical studies
Permanent slides of the pancreas were prepared and stained with eosin–hematoxylin for histopathological evaluation. Pancreatic tissue from all groups of animals was selectively taken from the gastro-spleenic portion and was Bouin’s fixed. Paraffin blocks were prepared, and 4–5 µm thin sections were cut with a rotary microtome, and routine microscopic slides were prepared. Hematoxylin and eosin-stained slides were microscopically (Primostar model, Carl Zeiss Meditec Group, Dublin, CA) examined by light for size and total number of cells present per islet by using Zeiss Axiovision LE imaging software (Carl Zeiss Meditec Group, Dublin, CA). The mean diameter of islet represents the mean of maximum diameter and a diameter at right angle to it (Mukherjee et al., Citation2004).
Determination of cytokine and CRP level
TNF-α and IL-6 levels in pancreatic tissue extract were estimated by using the ELISA kit obtained from Raybiotech (Norcross, GA). All samples were assayed in duplicate. The intra-assay variation was 6.7% and 5.5%, respectively, for TNF-α and IL-6. The Serum CRP level was also measured by using Rat CRP ELISA kit obtained from Raybiotech (Norcross, GA). The intra-assay variation was 3.4%. To avoid inter-assay variation, all samples were run at one time.
Immunohistochemistry
Pancreas sections of 5-µm thickness were deparaffinized, rehydrated in descending alcohol concentrations, and boiled in 10 mM sodium citrate (pH 6.0) for 30 min. Then, slides were maintained at sub-boiling temperature for 10 min and then reached to room temperature. They were then washed with distilled water for 5 min and incubated in 3% hydrogen peroxide for 10 min. Sections were incubated with PBS and then with blocking solution, normal goat serum in dilution 1/5 for 1 h. Sections without washing were incubated with the cleaved caspase-3 (Asp 175) antibody (Cell Signaling Technology Inc, Danvers, MA) at a dilution of 1/200 or NF-kB p65 antibody at a dilution of 1/1000 (Cell Signaling Technology Inc, Danvers, MA) overnight at 48 °C. Next day sections were washed thoroughly and incubated with the secondary antibody, goat anti-rabbit biotin conjugated (Sigma-Aldrich, St. Louis, MO), at a dilution of 1/300 for 30 min at room temperature. Sections were then incubated with extrAvidin peroxidase conjugate (Sigma, St. Louis, MO) for 30 min at room temperature at a dilution of 1/300 and stained with DAB tablets (Sigma, St. Louis, MO). After washing, sections were dehydrated in ascending alcohol concentrations, mounted, and examined under a light microscope.
Tunel assay
Apoptosis was estimated by the TUNEL assay, which relies on the incorporation of labeled dUTP at sites of DNA fragments. Sections of 5-µm thickness were deparaffinized and rehydrated in descending alcohol concentrations. Apoptotic cells in pancreatic tissue were detected using Apo-BrdU-IHC In situ DNA fragmentation assay kit obtained from BioVision, Milpitas, CA.
Statistics
Data were expressed as mean ± SE. Kruskal–Wallis non-parametric ANOVA test was performed to find whether or not scores of different groups differ significantly. To test intergroup significant difference, the Mann–Whitney U multiple comparison test was performed. Differences were considered significant if p < 0.05.
Results
To investigate the effects of nicotine on in vivo pancreatic islet function, the changes in fasting blood glucose, HbA1C level, and plasma insulin level in nicotine exposed rats were monitored. Chronic exposure to nicotine elevated fasting blood glucose level by 102.04% (p < 0.001). In the groups, where folic acid and folic acid with vitamin B12 were given orally along with nicotine injections, changes in fasting blood glucose was reversed (p < 0.05) and the values were significantly different from those of the only nicotine treated animals (). There was a significant decrease of 37.8% (p < 0.01) in the levels of insulin in nicotine-treated group which was insignificantly increased in the folic acid supplemented group by 23.36% (p > 0.05). The insulin level was almost the same in folic acid with vitamin B12-treated group (p < 0.05) (). The HbA1C level (), an objective index of chronic glycemia, was found to be increased significantly by 44.01% (p < 0.01) in nicotine-treated group compared with the control group. Supplementation of folic acid with or without vitamin B12 in other nicotine exposed rats reversed significantly the nicotine-induced changes in HbA1C level (p < 0.01).
Figure 1. Effect of folic acid (36 µg/kg body weight/d for 21 d) and folic acid + vitamin B12 (0.63 µg/kg body weight/d for 21 d) on nicotine (3 mg/kg body weight/d for 21 d)-induced changes in fasting blood glucose level. Data are expressed as mean ± SE. Significance level based on the Kruskal–Wallis test (p < 0.01)##. Control versus nicotine, p < 0.01**; nicotine versus nicotine + folic acid, p < 0.05*; nicotine versus folic acid + vitamin B12, p < 0.05*.
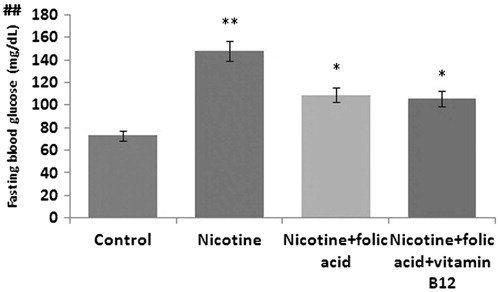
Figure 2. Effect of folic acid (36 µg/kg body weight/d for 21 d) and folic acid + vitamin B12 (0.63 µg/kg body weight/d for 21 d) on nicotine (3 mg/kg body weight/d for 21 d)-induced changes in [A] insulin level and [B] glycated hemoglobin level (HbA1C). Data are expressed as mean ± SE. Significance level based on the Kruskal–Wallis test (p < 0.01##, p < 0.001###). Control versus nicotine, p < 0.01**; nicotine versus nicotine + folic acid, p < 0.01** (insulin), p < 0.05* (HbA1C); nicotine versus folic acid + vitamin B12, p < 0.01** (insulin), p < 0.05* (HbA1C).
![Figure 2. Effect of folic acid (36 µg/kg body weight/d for 21 d) and folic acid + vitamin B12 (0.63 µg/kg body weight/d for 21 d) on nicotine (3 mg/kg body weight/d for 21 d)-induced changes in [A] insulin level and [B] glycated hemoglobin level (HbA1C). Data are expressed as mean ± SE. Significance level based on the Kruskal–Wallis test (p < 0.01##, p < 0.001###). Control versus nicotine, p < 0.01**; nicotine versus nicotine + folic acid, p < 0.01** (insulin), p < 0.05* (HbA1C); nicotine versus folic acid + vitamin B12, p < 0.01** (insulin), p < 0.05* (HbA1C).](/cms/asset/02b8d12e-265c-47c8-b665-5d5da4b2eee6/iphb_a_1043561_f0002_b.jpg)
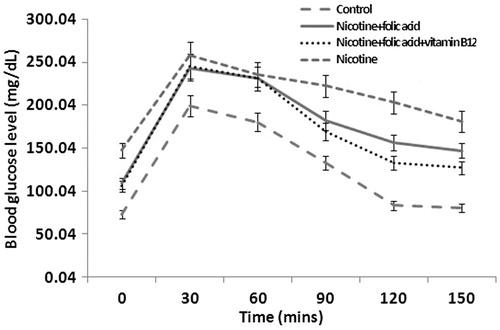
Further, to confirm that exposure to nicotine can cause islet damage resulting in blood glucose dysregulation, the oral glucose tolerance test (OGTT) was used. It was found that glucose tolerance was normal in control rats as indicated by the return of blood glucose level to nearly pre-glucose load level at 120 min after the glucose load (). In nicotine-treated rats, blood glucose attained peak at 30 min after glucose load (from 147.61 mg/dl to 257.94 mg/dl) but at 150 min, it returned only to 181.08 mg/dl (p < 0.05). Supplementation of folic acid alone or in combination with vitamin B12 was able to restore blood glucose by 71.62% (p < 0.01) and 84.63% (p < 0.01) at 150 min compared with its peak level at 30 min in nicotine-treated animals.
Figure 3. Effect of folic acid (36 µg/kg body weight/d for 21 d) and folic acid + vitamin B12 (0.63 µg/kg body weight/d for 21 d) on nicotine (3 mg/kg body weight/d for 21 d)-induced changes in oral glucose tolerance. Data are expressed as mean ± SE.
To further explore the involvement of oxidative stress damage in the mechanism underlying the toxicological effects induced by nicotine in the islets of rat, nitric oxide production, lipid peroxidation, glutathione content, and catalase and superoxide dismutase activity in pancreatic tissue homogenates were analyzed. Results revealed that MDA and NO levels, hallmarks of lipid peroxidation and inflammatory response, were markedly increased () in the nicotine-treated group. The levels of MDA and NO were increased by 40.71% (p < 0.01) and 63.08% (p < 0.01), respectively, in pancreatic tissue extract of the nicotine-treated group as compared with the control group. Folic acid alone or in combination with vitamin B12 treatment in other nicotine groups caused a marked decrease in both MDA level (p < 0.01) and NO production (p < 0.01).
Table 1. Effect of folic acid (36 µg/kg body weight/d for 21 d) and folic acid + vitamin B12 (0.63 µg/kg body weight/d for 21 d) on nicotine (3 mg/kg body weight/d for 21 d) induced changes in NO and MDA production, GSH level, and activity of SOD and CAT in pancreatic tissue of rat.
Also, chronic nicotine administration caused significant decrease (p < 0.01) in the tissue GSH levels of the pancreas as compared with the control group (). However, the nicotine-induced decrease in tissue GSH levels of pancreas were reversed by supplementation of folic acid only (p < 0.05) or folic acid with vitamin B12 (p < 0.01). In addition, after being intoxicated with nicotine, a decline in the activity of CAT (56.19%, p < 0.01) was observed in the nicotine-treated group when compared with the control group (). The CAT activity was restored to normal in folic acid and folic acid with vitamin B12-treated groups by 55.88% (p < 0.05), and 88.23% (p < 0.01), respectively. Furthermore, the SOD activity was compromised significantly (p < 0.01) by 29.16% in the nicotine-injected group while folic acid or folic acid with vitamin B12 supplementation in other nicotine-treated groups recovered by 78.54% (p < 0.01) and 83.27% (p < 0.01), respectively ().
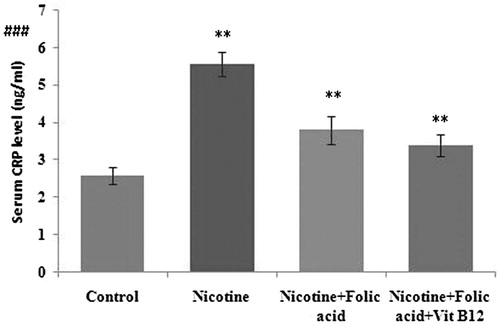
Cytokines are low-molecular-weight proteins released during the activation of the inflammatory cascade, which after binding to specific receptors affected immune cell differentiation, proliferation, and activity. Pro-inflammatory cytokines including TNF-α and IL-6 were quantified in this study. Compared with control, pancreatic TNF-α () was increased significantly in the nicotine-treated group, which was significantly ameliorated by folic acid supplementation (p < 0.01). Further supplementation of folic acid in combination with vitamin B12 also blunted the nicotine-induced changes in TNF-α production by 99.16% (p < 0.01). Similarly, pancreatic tissue IL-6 () was also increased significantly (37.33%, p < 0.01) in nicotine-treated rats as compared with control. Accordingly, folic acid alone or in combination with vitamin B12 blunted the nicotine-induced increase in IL-6 in pancreatic tissue extract (by 47.25% and 65.15%, respectively, p < 0.01). C-reactive protein (CRP), a circulating marker of systemic inflammation, was found to be significantly higher (p < 0.01) in the nicotine-exposed rat which was normalized to control level by both folic acid alone (p < 0.01) or folic acid and vitamin B12 (p < 0.01) ().
Figure 4. Effect of folic acid (36 µg/kg body weight/d for 21 d) and folic acid + vitamin B12 (0.63 µg/kg body weight/d for 21 d) on nicotine (3 mg/kg body weight/d for 21 d)-induced changes in [A] pancreatic tissue TNF-α [B] pancreatic tissue IL-6 level. Data are expressed as mean ± SE. Significance level based on the Kruskal–Wallis test (p < 0.001)###. Control versus nicotine, p < 0.01**; nicotine versus nicotine + folic acid, p < 0.01**; nicotine versus folic acid + vitamin B12, p < 0.01**.
![Figure 4. Effect of folic acid (36 µg/kg body weight/d for 21 d) and folic acid + vitamin B12 (0.63 µg/kg body weight/d for 21 d) on nicotine (3 mg/kg body weight/d for 21 d)-induced changes in [A] pancreatic tissue TNF-α [B] pancreatic tissue IL-6 level. Data are expressed as mean ± SE. Significance level based on the Kruskal–Wallis test (p < 0.001)###. Control versus nicotine, p < 0.01**; nicotine versus nicotine + folic acid, p < 0.01**; nicotine versus folic acid + vitamin B12, p < 0.01**.](/cms/asset/f4a843c2-bb71-47d3-abf3-6c7f68b2f949/iphb_a_1043561_f0004_b.jpg)
Figure 5. Effect of folic acid (36 µg/kg body weight/d for 21 d) and folic acid + vitamin B12 (0.63 µg/kg body weight/d for 21 d) on nicotine (3 mg/kg body weight/d for 21 d)-induced changes in serum CRP level. Data are expressed as mean ± SE. Significance level based on the Kruskal-Wallis test (p < 0.001)###. Control versus nicotine, p < 0.01**; nicotine versus nicotine + folic acid, p < 0.01**; nicotine versus folic acid + vitamin B12, p < 0.01**.
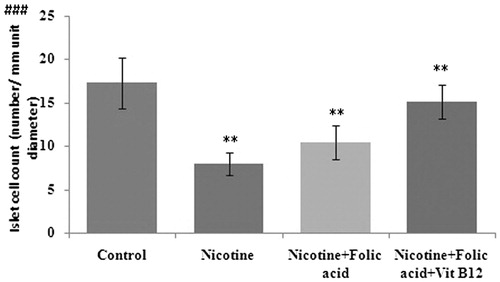
The light microscopic evaluation of the pancreas in the nicotine group showed degeneration of islets with vacuolization (). shows the effects of folic acid or folic acid with vitamin B12 supplementation on nicotine-induced changes in the population of islet cells of the rat pancreas. Results revealed that compared with control, in per unit diameter (µm) of islets, the number of islet cells in the nicotine-treated group was markedly low. Such reduction in islet cell counts was prevented by supplementation of folic acid alone or in combination with vitamin B12.
Figure 6. Representative photomicrograph of hematoxylin and eosin-stained section (×400) showing morphology and population of cells in pancreatic islet of [A] control, [B] nicotine (3 mg/kg body weight/d for 21 d), [C] folic acid (36 µg/kg body weight/d for 21 d), and [D] folic acid + vitamin B12 (0.63 µg/kg body weight/d for 21 d) supplemented rat. In [A], no pathological changes were seen. Normal architecture of pancreatic islet and surrounding acini was vivid. Islet size and cell population were normal. [B] Nicotine treated: typical characteristic of islet cell injury was present (marked by black arrow). Islet was shrunken; cell numbers were reduced. [C and D] Marked recovery of these cell injury changes by folic acid alone or in combination with vitamin B12.
![Figure 6. Representative photomicrograph of hematoxylin and eosin-stained section (×400) showing morphology and population of cells in pancreatic islet of [A] control, [B] nicotine (3 mg/kg body weight/d for 21 d), [C] folic acid (36 µg/kg body weight/d for 21 d), and [D] folic acid + vitamin B12 (0.63 µg/kg body weight/d for 21 d) supplemented rat. In [A], no pathological changes were seen. Normal architecture of pancreatic islet and surrounding acini was vivid. Islet size and cell population were normal. [B] Nicotine treated: typical characteristic of islet cell injury was present (marked by black arrow). Islet was shrunken; cell numbers were reduced. [C and D] Marked recovery of these cell injury changes by folic acid alone or in combination with vitamin B12.](/cms/asset/15fe2239-624a-413b-8e55-0ee07a14e692/iphb_a_1043561_f0006_b.jpg)
Figure 7. Effect of folic acid (36 µg/kg body weight/d for 21 d) and folic acid + vitamin B12 (0.63 µg/kg body weight/d for 21 d) on nicotine (3 mg/kg body weight/d for 21 d)-induced changes in islet cell count (no. per unit diameter). Significance level based on the Kruskal–Wallis test (p < 0.001)###. Control versus nicotine, p < 0.01**; nicotine versus nicotine + folic acid, p < 0.01**; nicotine versus folic acid + vitamin B12, p < 0.01**.
Activated NF-κB was assessed by immunocytochemistry by using activation-specific monoclonal antibodies to NF-κB p65 subunit, the epitope of which binds only after IκB dissociation. In control, the numbers of positively stained cells were low, whereas in the nicotine-treated group, NF-κB positive cells were readily observable (). Supplementation with only folic acid or folic acid and vitamin B12 in combination in nicotine-treated rats recovered nicotine-induced changes in NF-κB activation as evidenced by a decrease in the number of immunostained cells (). Similar patterns of results were also obtained in case of activated caspase 3 (). To investigate whether nicotine induced dysfunction of pancreatic islets via an apoptotic mechanism, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was performed. Results of our study revealed that there were higher numbers of TUNEL positive cells in nicotine-exposed rats (), while supplementation of folic acid alone or in combination with vitamin B12 was able to reduce the number of TUNEL positive cells in nicotine exposed rats.
Figure 8. NF-κB immunohistochemistry. Representative photomicrograph of pancreas section (×400) showing NF-κB expression in pancreatic islet of [A] control, [B] nicotine (3 mg/kg body weight/d for 21 d), [C] folic acid (36 µg/kg body weight/d for 21 d), and [D] folic acid + vitamin B12 (0.63 µg/kg body weight/d for 21 d) supplemented rat. Black arrow represents NF-κB positive cells.
![Figure 8. NF-κB immunohistochemistry. Representative photomicrograph of pancreas section (×400) showing NF-κB expression in pancreatic islet of [A] control, [B] nicotine (3 mg/kg body weight/d for 21 d), [C] folic acid (36 µg/kg body weight/d for 21 d), and [D] folic acid + vitamin B12 (0.63 µg/kg body weight/d for 21 d) supplemented rat. Black arrow represents NF-κB positive cells.](/cms/asset/9e35e542-a6f2-4cf6-a3a2-85abf816e7c4/iphb_a_1043561_f0008_b.jpg)
Figure 9. Caspase immunohistochemistry. Representative photomicrograph of pancreas section (×400) showing activated caspase expression in pancreatic islet of [A] control, [B] nicotine (3 mg/kg body weight/d for 21 d), [C] folic acid (36 µg/kg body weight/d for 21 d), and [D] folic acid + vitamin B12 (0.63 µg/kg body weight/d for 21 d)-supplemented rat. Black arrow represents caspase positive cells.
![Figure 9. Caspase immunohistochemistry. Representative photomicrograph of pancreas section (×400) showing activated caspase expression in pancreatic islet of [A] control, [B] nicotine (3 mg/kg body weight/d for 21 d), [C] folic acid (36 µg/kg body weight/d for 21 d), and [D] folic acid + vitamin B12 (0.63 µg/kg body weight/d for 21 d)-supplemented rat. Black arrow represents caspase positive cells.](/cms/asset/a629150e-789e-45ef-889b-1ee64430b5b5/iphb_a_1043561_f0009_b.jpg)
Figure 10. TUNEL assay. Representative photomicrograph of pancreas section (×400) showing tunel-positive cells in pancreatic islet of [A] control, [B] nicotine (3 mg/kg body weight/d for 21 d), [C] folic acid (36 µg/kg body weight/d for 21 d), and [D] folic acid + vitamin B12 (0.63 µg/kg body weight/d for 21 d)-supplemented rat. Black arrow represents tunnel positive cells.
![Figure 10. TUNEL assay. Representative photomicrograph of pancreas section (×400) showing tunel-positive cells in pancreatic islet of [A] control, [B] nicotine (3 mg/kg body weight/d for 21 d), [C] folic acid (36 µg/kg body weight/d for 21 d), and [D] folic acid + vitamin B12 (0.63 µg/kg body weight/d for 21 d)-supplemented rat. Black arrow represents tunnel positive cells.](/cms/asset/47301916-c699-47d5-a186-8d8faf7683e0/iphb_a_1043561_f0010_b.jpg)
Discussion
The present study reveals that nicotine causes pancreatic beta cell damage and promotes hyperglycemia in the rat model and that this effect is prevented by folic acid and vitamin B12. Results of this study also demonstrate the involvement of oxidative stress and increased production of proinflammatory cytokines and alteration in NF-κB expression as possible causes of injury and apoptosis in pancreatic islets of rats exposed to nicotine.
Earlier studies reported that nicotine need not act on surface nicotinic receptors (Borowitz & Isom, Citation2008) and that it is lipid soluble enough to penetrate into cells. Being an alkaloid, it is basic and would tend to collect in the relatively acidic intracellular environment. Also, intracellular organelles like endoplasmic reticulum or secretory granules have a pH of about 4, so nicotine would concentrate further inside these compartments. In addition, they also speculate that nicotine inhibits the release of insulin from isolated islets. Earlier, results support the hypothesis that smoking, and more specifically nicotine, leads to a persistent increase in blood glucose levels and as well as in HbA1C level (Clair et al., Citation2011). The present study provides evidence that nicotine induces pancreatic tissue oxidative stress in rats as compared with control animals, thus exacerbating islet cell damage, hyperglycemia, and impaired oral glucose tolerance test. In this study, a significant increase in fasting blood glucose and a decrease in plasma insulin levels in rats treated with nicotine (3 mg/kg body weight/d for 21 d i.p.) were observed. So, the results of higher blood glucose level and lower insulin level in nicotine-treated rats are consistent with the findings based on the human study (Borowitz & Isom, Citation2008; Clair et al., Citation2011). The oral glucose tolerance test is a widely used procedure in the diagnosis of diabetes and intermediate stages of hyperglycemia (Urita et al., Citation2006). Results indicate that after oral glucose administration, there was a marked difference in peak blood glucose level and blood glucose level at 2.30 h in control and nicotine-treated rats.
In addition, significantly higher HbA1C in nicotine-exposed rats as compared with control rats was observed. This hyperglycemic effect might be related to the decreased islet cell count in nicotine exposed rats as evidenced by the results of histological evaluation of hematoxylin–eosin-stained slides () of the pancreas. Islet cell count () was also found to be lower in nicotine-treated rats compared with control rats which might be accountable for lower insulin level and higher blood glucose and HBA1C level in rats treated with nicotine. On the basis of these results, it can be suggested that nicotine may be conscientious for the disturbance in blood glucose homeostasis; although the detailed mechanisms of this disturbance still require future investigation.
On one hand, earlier, increased free radical production and cooperative lipid peroxidation levels in various tissues of rats incubated with nicotine have been reported (Yildiz et al., Citation1999). On the other hand, oxidative stress is considered to play a role in the pathogenesis of different diseases, including cancer, diabetes, and cardiovascular diseases (Gul et al., Citation2000). Thus, it may be hypothesized that nicotine-induced oxidative stress may play an important role in the development of hyperglycemia and impaired glucose tolerance in smokers. To test this hypothesis, different oxidative stress parameters in pancreatic tissue of nicotine-exposed rats were measured. In the present study, marked elevation of lipid peroxidation and nitric oxide (NO) production in pancreatic tissue extract of nicotine-exposed rats indicates the enhanced generation of ROS (). Secondary lipid peroxidation products such as MDA may exert similar toxic effects, which can prolong and potentiate the primary free radical-initiated damage (Kalpana & Menon, Citation2004).
Since results of the present study demonstrated that oxidative stress takes part in the pathogenesis of nicotine-induced impairments in pancreatic islet cell functions, free radical ablation with the antioxidant agents seems to be beneficial for preventing the oxidant-induced tissue damage. Folic acid has been well defined as an effective free-radical scavenger and reported to inhibit lipid peroxidation (Blundell et al., Citation1996). Moreover, the anti-inflammatory effect of folic acid is manifested by a decrease in the levels of interleukin and C-reactive proteins. Folic acid deficiency lowers the concentrations of methyl tetrahydrofolate, the main methyl donor for methylation of homocysteine to methionine. Failures in remethylation also elevate the concentration of homocysteine (McDowell, Citation1989), which is reported to be high in smokers. Further, it has been reported that vitamin B12 and folic acid are required for methylation of homocysteine to methionine and vitamin B12 acts as methionine synthase enzyme to add the methyl group in the methylation process. In addition, antioxidant activity was reported to be more efficient when antioxidants were used in combination (Sahin et al., Citation2003).
In the study, supplementation of folic acid with vitamin B12 to nicotine-treated rats was found to be successful in reversing the nicotine-induced impairment in blood glucose homeostasis. These results were associated with increased islet beta cell mass and preserved islet architecture in nicotine-treated rats supplemented with folic acid alone or in combination with vitamin B12. Furthermore, in the present study, folic acid as a powerful antioxidant in combination with vitamin B12 inhibited NO production and MDA levels and potentiated SOD and CAT activities in pancreatic tissue extract of nicotine-exposed rats implicating that tissue integrity is maintained by inhibiting the breakdown of membrane phospholipids by lipid peroxidation (). These results are also well in line with earlier reports that oxidative stress plays a crucial role in inducing pancreatic islet β-cell injuries and the pathogenesis of diabetes mellitus, probably as a result of excessive levels of mitochondrial ROS production and the presence of fewer antioxidants enzymes in pancreatic β-cells (Drews et al., Citation2010; Rains & Jain, Citation2011).
Earlier reports have indicated that nicotine-induced toxicity is coupled with GSH depletion, which is one of the essential compounds for maintaining cellular integrity (El-Sokkary et al., Citation2007). In the present study, on one hand, nicotine exposure significantly depleted GSH stores in pancreatic tissue, indicating that GSH was used as an antioxidant for the detoxification of toxic oxygen metabolites, enhancing the susceptibility of the pancreatic islets to oxidative injury. On the other hand, folic acid and vitamin B12 reduced nicotine-induced oxidative injury with a concomitant maintenance of GSH stores in the pancreas, implicating its antioxidant action in improving the tissue functions. Overall, the findings on oxidant and antioxidant parameters suggest that nicotine-induced cytotoxicity on pancreatic islets is due to the generation of oxidative stress, which was ameliorated by the supplementation of folic acid in combination with vitamin B12.
In recent years, there has been a large number of studies, some of which are conflicting, in which, serum CRP concentrations have been measured in parallel to smoking status because of the possible link between smoking and the induction of inflammatory pathways (Yanbaeva et al., Citation2007). Earlier reports revealed that the CRP level was found to be significantly higher in male and female smokers compared with non-smokers, which could be considered reflecting an inflammatory episode (Lowe & Pepys, Citation2006). Also, baseline levels of C-reactive protein and interleukin-6 were significantly higher among those who subsequently developed diabetes compared with those who did not (Pradhan & Manson, Citation2001). In the present study, increased CRP level in nicotine-exposed rats signifies inflammatory state and islet cell destruction, which was counteracted by supplementation of folic acid and vitamin B12. The findings are well in line with the earlier finding based on the human study (Dietricha et al., Citation2007).
Several candidate inflammatory cytokines and factors such as IL-1β (interleukin 1β), INF-γ (interferon γ), TNF-α (tumor necrosis factor), oxygen-free radicals, and NO (nitric oxide) are implicated in promoting β-cell death with some synergistic effect (Mandrup-Poulsen, Citation2003). On the contrary, nicotine has been shown to increase the production of inflammatory mediators and being associated with pathogenesis of diseases by promoting proinflammatory mediators (Cooper & Magwere, Citation2008). In addition, it has been evidenced that smokers have a compromised antioxidant status and elevated concentrations of the TNF-α and IL-6 as a consequence of smoking, although nicotine has often implicated to be anti-inflammatory (Tappia et al., Citation1995). Therefore, in the present study, the effect of nicotine on cytokine production and the activation of NF-κB in pancreatic islets of rats were examined. It was found that nicotine can trigger the production of TNF-α and IL-6. Results revealed that nicotine-induced free radical generation and oxidative damage in pancreatic islets were associated with increased proinflammatory cytokine productions; which were ameliorated by combined supplementation of folic acid and vitamin B12 through decreased free radical generation. So, it is revealed that anti-inflammatory therapeutic approaches by folic acid and vitamin B12 perhaps directly or indirectly protect cells from the adverse effect of nicotine-induced oxidative damage in pancreatic islet cells.
IL-6 and IL-8 are important in the recruitment and the activation of inflammatory cells. The induction of these proinflammatory mediators is regulated by the activation of redox-sensitive transcription factor NF-κB (Rahman et al., Citation2003). NF-κB is a transcription factor which can be activated by oxidants and other stresses. Once activated, NF-κB translocates to the nucleus and binds to DNA and upregulates the expression of several genes such as iNOS and TNF-α. These target proteins initiate and propagate the immune/inflammatory response resulting in the invasion of immune cells into the pancreas that produces and amplifies the free radical production and destroys the beta cells ultimately (Ho et al., Citation2000). This may be the plausible explanation for nicotine-induced NF-κB activation and islet cell damage in our study. Combined supplementation of folic acid and vitamin B12 in nicotine-treated rats possibly inhibits the NF-κB activation by influencing the redox cycle state of the cell and reduces the severity of nicotine-induced pancreatic islet damage and impaired glucose homeostasis.
The apoptotic cysteine protease, caspase-3, is normally expressed as a 32-kDa precursor, but following apoptosome formation by cytochrome c, the protein is cleaved, first into p20 and p12 fragments and then the p20 subunit is further proteolyzed to form the mature 17-kDa fragment (Han et al., Citation1997). The active form of caspase-3 is classified as an ‘‘executioner’’ caspase and is responsible for the majority of cellular apoptotic events (Zimmermann et al., Citation2001). Hence, active caspase-3 was selected as a representative protein for the localization of apoptotic signaling within the pancreas on exposure to nicotine. Immunopositive staining of the active executioner caspase in nicotine-treated animals suggest apoptosis induction in islet cells. Results suggest that folic acid in combination with vitamin B12 has an ameliorating influence to help prevent the nicotine-induced apoptosis in pancreatic islet cell.
Conclusion
In conclusion, supplementation with folic acid and vitamin B12 was found to reduce hyperglycemia and pancreatic islet cell damage by modulating oxidative stress, cytokine production, and apoptosis in nicotine-treated rats. One mechanism may be through its scavenging ability to protect the pancreatic islets from free radicals-damage induced by nicotine in vivo.
The findings suggest that folic acid and vitamin B12 can be used as a nutritional supplement, especially by people who smoke, in order to prevent nicotine-induced toxicity in pancreatic islets.
Acknowledgements
The authors thank Mr. Sumantra Kumar Ghoshal for his kind help in preparing histological slides.
Declaration of interest
The authors report no declarations of interest. The authors are grateful to University Grants Commission (UGC), Government of India for funding this study [Sanction no. 41-72/2012 (SR)] as Major Research Project.
References
- Aebi H. (1984). Catalase in vitro. Methods Enzymol 105:121–6
- Attvall S, Fowelin J, Lager I, et al. (1993). Smoking induces insulin resistance – A potential link with the insulin resistance syndrome. J Intern Med 233:327–32
- Balfour D, Benowitz N, Fagerstrom K, et al. (2000). Diagnosis and treatment of nicotine dependence with emphasis on nicotine replacement therapy: A status report. Eur Heart J 21:438–45
- Bell D. (2010). Metformin-induced vitamin B12 deficiency presenting as a peripheral neuropathy. South Med J 103:265–7
- Blundell G, Jones BG, Rase FA, et al. (1996). Homocysteine mediated endothelial cell toxicity and its amelioration. Atherosclerosis 122:163–72
- Borowitz JL, Isom GE. (2008). Nicotine and type II diabetes. Toxicol Sci 103:225–7
- Chattopadhyay K, Chattopadhyay BD. (2008). Effect of nicotine on lipid profile, peroxidation and antioxidant enzymes in female rats with restricted dietary protein. Indian J Med Res 127:571–6
- Clair C, Bitton A, Meigs JB, et al. (2011). Relationships of cotinine and self-reported cigarette smoking with hemoglobin A1c in the U.S. Diabetes Care 34:2250–5
- Cooper RG, Magwere T. (2008). Nitric oxide-mediated pathogenesis during nicotine and alcohol consumption. Indian J Physiol Pharmacol 52:11–18
- Daniel M, Cargo MD. (2004). Association between smoking, insulin resistance and beta-cell function in a North-Western First Nation. Diabetic Med 21:188–93
- Das S, Neogy S, Gautam N, et al. (2009). In vitro nicotine induced superoxide mediated DNA fragmentation in lymphocytes: Protective role of Andrographis paniculata Nees. Toxicol In Vitro 23:90–8
- Dietricha T, Garciaa RI, de Pablob P, et al. (2007). The effects of cigarette smoking on C-reactive protein concentrations in men and women and its modification by exogenous oral hormones in women. Eur J Cardiovasc Prev Rehabil 14:694–700
- Drews G, Krippeit-Drews P, Dufer M. (2010). Oxidative stress and β-cell dysfunction. Pflugers Arch 460:703–18
- El-Sokkary GH, Cuzzocrea S, Reiter RJ. (2007). Effect of chronic nicotine administration on the rat lung and liver: Beneficial role of melatonin. Toxicology 239:60–7
- Facchini FS, Hollenbeck CB, Jeppesen J, et al. (1992). Insulin resistance and cigarette smoking. Lancet 339:1128–30
- Gul M, Kutay FZ, Temocin S, et al. (2000). Cellular and clinical implications of glutathione. Indian J Exp Biol 38:625–34
- Gupta PC. (2001). The public health impact of tobacco. Curr Sci 8:475–81
- Han Z, Hendrickson EA, Bremner TA, et al. (1997). A sequential two-step mechanism for the production of the mature p17:p12 form of caspase-3 in vitro. J Biol Chem 27:13432–6
- Hermann L, Nilsson B, Wettre S. (2004). Vitamin B12 status of patients treated with metformin: A cross-sectional cohort study. Br J Diabetes Vasc Dis 4:401–6
- Ho E, Chen G, Bray T. (2000). Alpha-phenyl-tert-butylnitrone (PBN) inhibits NF-κB activation offering protection against chemically induced diabetes. Free Radic Biol Med 4:604–14
- Joshi R, Adhikari S, Patro BS, et al. (2001). Free radical scavenging behavior of folic acid: Evidence for possible antioxidant activity. Free Radic Biol Med 30:1390–9
- Kalpana C, Menon VP. (2004). Modulatory effects of curcumin on lipid peroxidation and antioxidant status during nicotine-induced toxicity. Pol J Pharmacol 56:581–6
- Khan NA. (2002). Antioxidant and cancers. Sciences 2:38–43
- Liu K, Dai L, Jean W. (2006). Metformin-related vitamin B12 deficiency. Age Ageing 35:200–1
- Liu Z, Wang J, Yang Z, et al. (2013). Antioxidative effects of hesperetin against lead acetate-induced oxidative stress in rats. Ind J Pharmacol 45:395–8
- Lortz S, Tiedge M. (2003). Sequential inactivation of reactive oxygen species by combined overexpression of SOD isoforms and catalase in insulin-producing cells. Free Radic Biol Med 34:683–8
- Lowe GD, Pepys MB. (2006). C-reactive protein and cardiovascular disease: Weighing the evidence. Curr Atheroscler Rep 8:421–8
- Lowry OH, Rosebrough NJ, Farr AL, et al. (1961). Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–75
- Luqman S, Rizvi SI. (2006). Protection of lipid peroxidation and carbonyl formation in proteins by capsaicin in human erythrocytes subjected to oxidative stress. Phytother Res 20:303–6
- Mahapatra SK, Chakraborty SP, Roy S. (2010). In vitro time dependent nicotine-induced free radical generation and status of glutathione cycle in murine peritoneal macrophage. Al Ameen J Med Sci 3:182–94
- Mandrup-Poulsen T. (2003). Apoptotic signal transduction pathways in diabetes. Biochem Pharmacol 66:1433–40
- McCarty MF. (2000). Increased homocysteine associated with smoking, chronic inflammation and aging may reflect acute-phase induction of pyridoxal phosphatase activity. Med Hypotheses 55:289–93
- McDowell LR. (1989). Vitamins in Animal Nutrition, Vitamin C, Folacin: Comparative Aspects to Human Nutrition. London: Academic Press, 298–322
- Momi N, Kaur S, Ponnusamy MP, et al. (2012). Interplay between smoking-induced genotoxicity and altered signaling in pancreatic carcinogenesis. Carcinogenesis 33:1617–28
- Mukherjee S, Das D, Darbar S, et al. (2004). Arsenic trioxide generates oxidative stress and islet cell toxicity in rabbit. Curr Sci 86:854–7
- Mukherjee S, Das D, Mukherjee M, et al. (2006). Synergistic effect of folic acid and vitamin B12 in ameliorating arsenic-induced oxidative damage in pancreatic tissue of rat. J Nutr Biochem 17:319–27
- Muthukumaran S, Sudheer AR, Menon VP, et al. (2008). Protective effect of quercetin on nicotine-induced prooxidant and antioxidant imbalance and DNA damage in Wistar rats. Toxicology 243:207–15
- Muzawar SA, Patil VW. (2011). Folic acid and vitamins B12 levels and their correlation in cigarette smoker with hyperhomocysteinemia. Al Ameen J Med Sci 4:169–74
- Mysore TB, Shinkel TA, Collins J, et al. (2005). Overexpression of glutathione peroxidase with two isoforms of superoxide dismutase protects mouse islets from oxidative injury and improves islet graft function. Diabetes 54:2109–16
- Neogy S, Das S, Kar Mahapatra S, et al. (2008). Amelioratory effect of Andrographis paniculata Nees on liver, kidney, heart, lung and spleen during nicotine induced oxidative stress. Environ Toxicol Pharmacol 25:321–8
- Nervo M, Lubini A, Raimundo F, et al. (2011). Vitamin B12 in metformin-treated diabetic patients: A cross-sectional study in Brazil. Rev Assoc Med Bras 57:46–9
- Okuyemi KS, Harris J, Ahuwalia JS, et al. (2001). Documentation of smoking: Role of age, gender and ethnicity. J Assoc Minor Acad Physicians 12:125–8
- Pagan K, Hou J, Goldenberg RL, et al. (2001). Effect of smoking on serum concentrations of total homocysteine and B vitamins in mid-pregnancy. Clin Chim Acta 306:103–9
- Pflipsen M, Oh R, Saguil A, et al. (2009). The prevalence of vitamin B12 deficiency in patients with Type 2 diabetes: A cross-sectional study. J Am Board Fam Med 22:528–34
- Pradhan AD, Manson JE. (2001). C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 286:327–34
- Radzeviciene L, Ostrauskas R. (2009). Smoking habits and the risk of type 2 diabetes: A case control study. Diabetes Metab 35:192–7
- Rahman I, Gilmour PS, Jimenez LA, et al. (2003). Ergothioneine inhibits oxidative stress and TNF-α-induced NF-kappa B activation and interleukin-8 release in alveolar epithelial cells. Biochem Biophys Res Commun 302:860–4
- Rains JL, Jain SK. (2011). Oxidative stress, insulin signaling, and diabetes. Free Radic Biol Med 50:567–75
- Raso GM, Meli R, Gualillo O, et al. (1999). Prolactin induction of nitric oxide synthase in rat C6 Glioma cells. J Neurochem 73:2272–7
- Reis RP, Azinheira J, Reis HP, et al. (2000). Influence of smoking on homocysteinemia at baseline and after methionine load. Rev Port Cardiol 119:471–4
- Sahin K, Onderci M, Sahin N, et al. (2003). Dietary vitamin c and folic acid supplementation ameliorates the detrimental effects of heat stress in Japanese Quail. J Nutr 133:1882–6
- Sedlak J, Lindsay RH. (1968). Estimation of total protein bound and nonprotein sulfhydryl groups in tissue with Ellman’s reagent. Ann Clin Biochem 25:192–205
- Sun Y, Oberley LW, Li YA. (1988). A simple method for clinical assay of superoxide dismutase. Clin Chem 34:497–500
- Tappia PS, Troughton KL, Langley-Evans SC, et al. (1995). Cigarette smoking influences cytokine production and antioxidant defences. Clin Sci (Lond) 88:485–9
- Urita Y, Ishihara S, Akimoto T, et al. (2006). Seventy five gram glucose tolerance test to assess carbohydrate malabsorption and small bowel bacterial overgrowth. World J Gastroenterol 12:3092–5
- Valenca SS, Gouveia L, Pimenta WA, et al. (2008). Effects of oral nicotine on rat liver stereology. Int J Morphol 26:1013–22
- Wills ED. (1987). Evaluation of lipid peroxidation in lipids and biological membranes. In: Snell K, Mullock B, eds. Biochemical Toxicology: A Practical Approach. Oxford (England): IRL Press, 138–40
- Yanbaeva DG, Dentener MA, Creutzberg EC, et al. (2007). Systemic effects of smoking. Chest 131:1557–66
- Yeh HC, Duncan BB, Schmidt MI, et al. (2010). Smoking, smoking cessation and risk for type 2 diabetes mellitus. Ann Intern Med 152:10–17
- Yildiz D, Liu YS, Ercal N, et al. (1999). Comparison of pure nicotine and smokeless tobacco extract-induced toxicities and oxidative stress. Arch Environ Contam Toxicol 37:434–9
- Yoshikawa H, Hellstrom-Lindahl E, Grill V. (2005). Evidence for functional nicotinic receptors on pancreatic beta cells. Metabolism 54:247–54
- Zimmermann KC, Bonzon C, Green DR. (2001). The machinery of programmed cell death. Pharmacol Ther 92:57–70