
Abstract
Context: Bisbibenzyl compounds have gained our interests for their potential antitumor activity in malignant cell-types.
Objective: The objective of this study is to investigate the effect of bisbibenzyl compounds riccardin C (RC), marchantin M (MM), and riccardin D (RD) on androgen receptor (AR) in prostate cancer (PCa) cells.
Materials and methods: After exposure to 10 μM of the compounds for 24 h, cell cycle and cell survival analyses were performed using FACS and MTT assay to confirm the effect of these bisbibenzyls on PCa LNCaP cells. Changes in the AR expression and function, as the result of exposure to the compounds, were investigated using real-time PCR, ELISA, transient transfection, western blotting (WB), immunoprecipitation, and immunofluorescence staining (IF). Chemical-induced autophagy was examined by WB, IF, and RNAi.
Results: RC, MM, and RD reduced the viability of LNCaP cells accompanied with arrested cell cycle in the G0/G1 phase and induction of apoptosis. Further investigation revealed that these compounds significantly inhibited AR expression at mRNA and protein levels, leading to the suppression of AR transcriptional activity. Moreover, inhibition of proteasome activity by bisbibenzyls, which in turn caused the induction of autophagy, as noted by induction of LC3B expression, conversion, and accumulation of punctate dots in treated cells. Co-localization of AR/LC3B and AR/Ub suggested that autophagy contributed to the degradation of polyubiquitinated-AR when proteasome activity was suppressed by the bisbibenzyls.
Discussion and conclusion: Suppression of proteasome activity and induction of autophagy were involved in bisbibenzyl-mediated modulation of AR activities and apoptosis, suggesting their potential in treating PCa.
Introduction
Prostate cancer (PCa), the most common cancer diagnosed in men, is a hormonally driven disease. Since the development of PCa is clearly linked to androgen stimulation of the androgen receptor (AR) system, androgen ablation therapy is an effective treatment for advanced PCa through inhibition of AR activity. However, it still remains highly malignant because PCa relapses to be castration-resistant prostate cancer (CRPC) that is resistant to further hormonal depletion therapy (Feldman & Feldman, Citation2001). Despite increased public awareness and improved strategies for intervention of the tumors, there are no effective treatments for patients with metastatic CRPC due to complicated mechanisms underlying the development of this disease (Zhu & Kyprianou, Citation2008) as revealed in clinical investigations. Compelling evidence has demonstrated that the AR remains inappropriately activated at castration levels of androgens, via multiple mechanisms including the crosstalk between the AR and elevated oncogenic survival pathways of receptor tyrosine kinases, suggesting pro-survival effect mediated by the AR signaling is a key contributor to CRPC development (Seruga et al., Citation2011; Zhu & Kyprianou, Citation2008). Therefore, developing effective ways to block AR activity and eliminating the abundance of AR are still of curative strategies for PCa (Bluemn & Nelson, Citation2012).
The expression and the transcriptional activity of AR are modulated by a variety of factors including transcription factors, RNA-binding proteins, and coregulators of AR (Heinlein & Chang, Citation2002). For example, AR levels are upregulated by Sp1 and cAMP response element-binding protein at transcriptional level. HuR protein is implicated in the regulation of AR post-transcriptionally (Yeap et al., Citation2002). The increased quantities of AR protein may also be caused by prolonged stability and slower turnover. Interaction between AR and coregulators, such as steroid receptor coactivators (SRC-1), or various proteins that control post-translational modifications including phosphorylation, sumoylation, ubiquitination, and acetylation, can result in increased AR activity (Heinlein & Chang, Citation2002). Thus, the complex regulation of AR activity in PCa may provide new optional strategies for the management of the disease achieved via controlling AR expression and function by multiple mechanisms.
The 26S proteasome is an enzyme complex containing multiple catalytic units that degrade polyubiquitinated proteins. There are three major proteasome activities: chymotrypsin-like (CT-like), trypsin-like, and peptidyl–glutamyl peptide hydrolyzing (PGPH) activities (Loidl et al., Citation1999). Compelling evidence indicates that the ubiquitin/proteasome system, a major pathway mediating intracellular protein turnover, also participates in the regulation of gene expression and function and is associated with tumor cell survival (Kar et al., Citation2013; Micel et al., Citation2013). Therefore, proteasomal degradation of regulatory proteins in cells is an effective mechanism for regulating cellular processes, including cell survival and cell death. It has been demonstrated that AR expression is controlled post-translationally by the ubiquitin/proteasome system either in androgen-dependent or -independent manner, where the latter occurs in response to growth factors and cytokine stimulation. In addition, the subunit of proteasome is an essential element of the AR transcriptional complex located on target gene promoters, and inhibition of the proteasome decreases AR transcriptional activity (Chymkowitch et al., Citation2011; Jaworski, Citation2006). Thus, chemical inhibitors of the proteasome have emerged as effective antitumor therapies (Adams, Citation2004; Kar et al., Citation2013). Bortezomib (Velcade/PS-341), a peptide boronate inhibitor of the proteasome, is currently approved for clinical use to treat multiple myeloma and mantle cell lymphoma. Many efforts are being made to develop numerous structurally distinct proteasome inhibitors including naturally occurring compounds (Chen et al., Citation2007; Mori et al., Citation2006; Yang et al., Citation2007). For example, clioquinol is able to inhibit the proteasomal CT-like activity associated with repression of AR expression, and induces apoptosis in PCa cells (Chen et al., Citation2007). Inhibition of CT-like activity of proteasome by a natural compound withaferin A results in loss of AR and activation of caspase-3 in PCa cells (Yang et al., Citation2007).
Most recently, macrocyclic bisbibenzyl compounds found in liverwort plants have gained our interest for their potent antitumor activities in malignant cell-types (Shi et al., Citation2008, Citation2009; Xu et al., Citation2010, Citation2012). However, the detailed molecular mechanisms and targets underlying their activities remain unclear. We have recently identified inhibitory effect of marchantin M (MM) on proteasome activity, leading to the activation of autophagy in PCa cells (Jiang et al., Citation2013). This current work was designed to investigate if riccardin C (RC) (Lu et al., Citation2006; Xu et al., Citation2010), MM (Qu et al., Citation2007; Xu et al., Citation2010), and riccardin D (RD) (Lu et al., Citation2006; Xue et al., Citation2012), which induced apoptosis in malignant PCa cells are able to inhibit AR activity, a key molecule in modulating androgen-dependent human PCa LNCaP cell survival. The LNCaP cell line harboring the AR with single missense mutation on ligand-binding lesions at Thr877Ala has been extensively studied as a cell model for androgen responsive research. Our results displayed that these three compounds abrogated AR expression and transcriptional function. Suppression of proteasome activity and induction of autophagy were involved in bisbibenzyl-mediated modulation of AR activities and apoptosis in PCa cells.
Materials and methods
Chemicals and reagents
The macrocyclic bisbibenzyls riccardin C (Lu et al., Citation2006), riccardin D (Lu et al., Citation2006), and marchantin M (Qu et al., Citation2007) were isolated from Dumortiera hirsuta, and Asterella angusta. Their structural determination and purity were described previously (Lu et al., Citation2006; Qu et al., Citation2007). These compounds were dissolved in dimethyl sulfoxide (DMSO) and stored as small aliquots at −20 °C. A synthetic androgen, mibolerone (Mib, New England Nuclear, Boston, MA) was dissolved in ethanol.
Cell culture and treatments
Human PCa cell lines LNCaP (The American Type Culture Collection, Rochville, MD) and PC-3 (The Cell Bank of Chinese Academy of Sciences, Shanghai) were seeded in 75 ml flasks or 24-well plates and cultured in RPMI 1640 medium (HyClone, Logan, UT) supplemented with 10% fetal bovine serum (FBS) (HyClone, Logan, UT) and 5% CO2 at 37 °C until they reached approximately 60–70% confluence. LNCaP cells were maintained in serum-free RPIM 1640 medium for 24 h to deplete endogenous steroid hormones prior to treatment with the bisbibenzyl chemicals at desired concentrations with or without 1 nM of Mib in the medium containing charcoal stripped serum. Ethanol and DMSO were used as control vehicles.
Cell viability assay
LNCaP cells were propagated in 24-well plates and treated with vehicle, Mib alone or Mib combined with desired concentrations of the chemicals for 24 h. Changes in cell viability in response to the chemicals were examined by 3-(4,-5-dimethylthiazol-2-yl)-2,-5-diphenyl-2H-tetrazolium bromide (MTT, Sigma, St. Louis, MO) colorimetric assays. After incubation with 10 μl MTT for 4 h, the cell growth response was detected by measuring the absorbance at 570 nm on a plate reader (Bio-Rad, Hercules, CA). Three replicates were used for each treatment.
Cell cycle and apoptosis analysis
LNCaP cells were cultured in 75 ml flasks under the conditions described above. After exposure to the chemicals for 24 h, cells were collected, washed with ice-cold phosphate buffered saline (PBS), and then fixed in 70% ethanol. Cell cycle and apoptosis analyses were determined with propidium iodide (PI) staining and flow cytometry using the Becton Dickinson FACScan (Becton Dickinson, Franklin Lakes, NJ).
Transient transfection assay
Cells were seeded in 24-well plates and grown under the conditions described above. LNCaP cells were transfected with a luciferase reporter pGL3 basic vector with the PSA-6kb promoter (PSA promoter, 1 μg/well), or AR-2kb promoter (AR promoter, 1 μg/well), or a pGL3-SV40 construct with three copies of androgen response element (ARE) in the hk2 promoter (hk2-3ARE, 1 μg/well) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The empty vector pGL3 basic (1 μg/well) and pGL3-SV40 (1 μg/well) were used as controls. The phRL-TK vector (0.05 μg/well) was co-transfected to normalize transfection efficiency. For transfection into PC-3 cells, human AR expression vector pSG5-AR (0.5 μg/well) was co-transfected with desired plasmids. After 24 h of transfection, cells were exposed to the chemicals in the presence or absence of Mib (1 nM) for an additional 24 h. Untreated cells served as a control. Cell lysates were prepared for luciferase assays (Dual-luciferase reporter assay system, Promega, Madison, WI). The ratio of promoter- or ARE-mediated luciferase activities to phRL-TK luciferase activities represents relative luciferase activities. The results were taken from three independent experiments.
Quantitative PCR
After exposure to the chemicals for 24 h, LNCaP cells were collected and total RNA was extracted using Trizol (Invitrogen, Carlsbad, CA). AR transcript was detected from M-MLV reverse transcriptase-amplified cDNA, with one aliquot designated to receive no enzyme as a control. Quantitative PCR (TaqMan PCR) was performed using Premix Ex TaqTM reagent (Takara, Berkeley, CA) on the Takara Thermal Cycle Dice TP800 Detection System (Takara, Berkeley, CA). Changes in AR gene expression after treatment were quantified as previously described (Yuan et al., Citation2008).
PSA quantification assay
LNCaP cells were exposed to the chemicals in the presence or absence of Mib for 24 h where untreated cells served as a control. PSA protein level in spent medium was determined with ELISA assay kit (Roche, Nutley, NJ) based on the instructions of the manufacturer.
Proteasome activity assay
The chymotrypsin-like (CT-Like) activity of proteasome was measured by incubation of peptide substrate Suc–Leu–Leu–Val–Tyr–AMC (Suc-LLVY-AMC) with either cell lysates from chemical-treated cells or with purified 20S proteasome. The substrate was incubated with 20S proteasome at 37 °C for 40 min in the presence or absence of chemicals in assay buffer, and then the enzyme activity was evaluated by the production of hydrolyzed AMC groups with an excitation of 380 nm and an emission of 460 nm. To measure the CT-Like activity of proteasome in whole cell lysates, cells were exposed to 10 μM of the chemicals. Cell lysates were prepared with lysis buffer (50 mM Tris HCl, pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.5% NP40, and 2 mM DTT), and protein concentration was quantified with Bradford assay. Aliquots of 20 μg protein were incubated with the substrate at 37 °C for 40 min. Fluorescent activity of hydrolyzed AMC groups was measured using a Mithras LB-940 (Berthold Technologies, Bad Wildbad, Germany).
Western blotting
After treatment of cells with desired chemicals in the presence or absence of Mib for 24 h, cells lysates were prepared using RIPA buffer containing fresh protease inhibitor mixture (50 μg/ml aprotinin, 0.5 mM phenylmethanesulfonyl fluoride, 1 mM sodium orthovanadate, 10 mM sodium fluoride and 10 mM β-glycerolphosphate). Nuclear extracts from LNCaP cells with or without pretreatment with the chemicals for 24 h were prepared as described previously (Jiang & Eberhardt, Citation1995). Proteins were quantified using the BCA protein assay (Biocolor BioScience & Technology, Shanghai, China). Blots were incubated with specific antibodies against Bcl-2, Bax (Cell Signaling Technology, Danvers, MA), AR (BD Biosciences, San Jose, CA), histone (Cell Signaling Technology, Danvers, MA), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), SRC-1, ubiquitin (Ub), p21Cip1, HA, or β-tubulin (Santa Cruz Technology, Dallas, TX). Visualization of the immunocomplexes was done by enhanced chemiluminescence detection system (Millipore, Huntington Beach, CA) and followed by exposure to X-ray films. For the detection of polyubiquitinated-AR proteins, we transfected an Ub expression plasmid with HA tag (a gift kindly provided by Professor Chengjiang Gao, Shandong University School of Medicine) into LNCaP cells. After 24 h transfection, the cells were treated with the chemicals as indicated for an additional 24 h, and then the cell lysates were prepared for western blot assay.
Co-immunoprecipitation
LNCaP cells were transfected with the HA-Ub expression plasmid for 24 h, and then exposed to the chemicals for an additional 24 h. Whole cell lysates were prepared and precleared with anti-rabbit IgG and protein A-agarose (Santa Cruz Technology, Dallas, TX). Aliquots of 500 μg precleared proteins were incubated with 1 μg of antibody against AR or HA in binding buffer (20 mM HEPES, pH 7.9, 20% glycerol, 150 mM KCl, 0.2 mM EDTA, 0.5 mM dithiothreitol, 0.5 mM PMSF, 50 μg/ml aprotinin, 1 mM sodium orthovanadate, 10 mM sodium fluoride, and 10 mM β-glycerolphosphate) overnight at 4 °C, followed by the addition of protein A beads for 4 h at 4 °C. The immunoprecipitates were washed with buffer containing 50 mM Tris-HCl, pH 7.5, 0.5% IGEPAL CA-630, 150 mM NaCl, 0.2 mM EDTA, 0.5 mM PMSF, 50 μg/ml aprotinin, 1 mM sodium orthovanadate, 10 mM sodium fluoride, and 10 mM β-glycerolphosphate. Immunocomplexes were recovered by heating at 75 °C for 10 min in SDS sample buffer and analyzed by Western blot assay.
Immunofluorescence staining
Cells were grown on coverslips. After incubation with the chemicals for 24 h, the cells were fixed with ice-cold methanol/acetone (1:1) and incubated with 3% BSA in PBS with 0.1% Triton X-100. The cells were then incubated with primary antibodies, rinsed with PBS, then immunostained with secondary antibodies, and finally stained with DAPI (2 mg/ml). Fluorescence images were captured using a confocal microscopy (Carl Zeiss, Dublin, CA).
GFP-LC3 assay
GFP-LC3B vector (1 µg) was transfected into 2 × 105 cells for 24 h, then exposed to chemicals for an additional 24 h. The fluorescence images were then scored under a Nikon phase-fluorescence microscope (Nikon Instruments, Cedar Grove, NJ).
RNA interference
LNCaP cells were incubated in 6-well plates and transfected with siRNA oligonucleotides using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Chemically modified siRNAs targeting ATG5i and negative control NCi siRNA were purchased from Invitrogen (Carlsbad, CA). After 24 h transfection, cells were treated with the chemicals or vehicle in the presence or absence of Mib (1 nM) for an additional 24 h, and cell lysates were subjected to western blot assay. Cell viability and cell apoptosis following treatment were determined by MTT and PI exclusion assays as described above.
Statistical analysis
The data are presented as mean ± SD of several independent experiments. Statistical analysis was performed using 2-tailed Student's t-test for paired values, and statistical significance was considered when a p was < 0.05.
Results
Bisbibenzyls induce LNCaP cell-cycle arrest and apoptosis
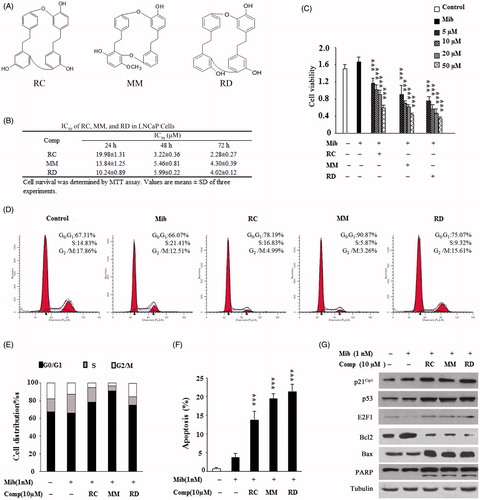
Given the fact that dysregulation of AR in PCa cells plays an important role in cell proliferation and survival, we chose LNCaP cell line-expressing mutant AR as a model to test whether the bisbibenzyls affect AR activity. Initially, we verified the inhibitory effect of these agents on LNCaP cell proliferation. As shown in , compounds RC, MM, and RD dose dependently reduced the viability of LNCaP cells. The IC50 values of RC, MM, and RD were 19.9, 13.8, and 10.2 μM, respectively. As cell growth inhibition is often related to perturbation with cell cycle progression, the results in displayed that RC, MM, and RD arrested LNCaP cells in the G0/G1phase in the presence of Mib compared with the Mib-treated cells alone, correlating with decreased S-phase. Accordingly, bisbibenzyl treatments increased the expression of p53, p21Cip1, and E2F-1 that are the molecular markers correlated with G0/G1 phase arrest (). Of note, Mib treatment suppressed E2F-1 expression while these compounds moderately rescued E2F-1 expression in the presence of Mib, consistent with the observations that androgen downregulates E2F-1 expression in the presence of AR (Huang et al., Citation2004). Moreover, cell-cycle deregulation leading to apoptosis was noticeably observed when LNCaP cells were treated with RC, MM, and RD as indicated by accumulation of the apoptotic sub-G1 population (). As summarized in , induction of apoptosis by RC, MM, and RD was 13.7, 19.4, and 21.3% at 10 μM concentration, respectively, consistent with our previous observations that bisbibenzyls inhibited cell proliferation and induced apoptosis in PC3 cells (Xu et al., Citation2010). Meanwhile, impairment of Bcl-2, accumulation of Bax, and the cleaved PARP, which are hallmarks for apoptotic response, were detectable in the chemical-treated cells (). Thus, these results support the notion that RC, MM, and RD exhibited inhibitory effect on LNCaP cells by cell-cycle arrest and induction of apoptosis, where MM and RD were somewhat more potent than RC.
Figure 1. Analysis of growth inhibitory effect of RC, MM, and RD on LNCaP cells. (A) Chemical structures of RC, MM, and RD. (B) Summary of IC50 values of RC, MM, and RD in LNCaP cells. (C) The effect of 24 h treatment of RC, MM, and RD on growing LNCaP cells was determined by MTT assay. Control cells were treated with equal volumes of DMSO supplemented in medium. ***p < 0.001 compared with the Mib+ control group. (D) After exposure of LNCaP cells to RC, MM, or RD for 24 h, cells were harvested, and stained with propidium iodide, and cell cycle was analyzed by flow cytometry. (E) Percentage of cell populations in cell cycle. (F) Percentage of apoptotic cells. ***p < 0.001 compared with the Mib+ control group. (G) Western blotting analysis of p53, p21Cip1, E2F-1, Bcl2, Bax, and PARP in the cells after RC, MM, or RD treatment for 24 h.
Bisbibenzyls suppress AR expression with reduced ligand-induced AR nuclear translocation and transactivation
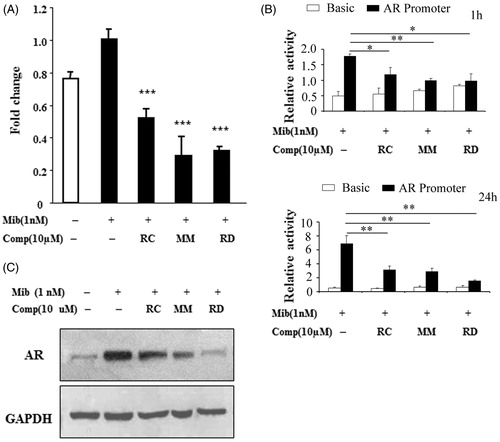
We next investigated if RC, MM, and RD affect AR expression in LNCaP cells, which in turn leads to cellular apoptosis. As demonstrated by quantitative PCR analysis, endogenous AR mRNA was increased by Mib treatment, while Mib-mediated stimulation in the AR expression was significantly repressed in cells exposed to RC, MM, and RD (). We also determined the effect of the bisbibenzyls on AR promoter activity using luciferase activity assays. As shown in , androgen-induced transcription was evident in cells transfected with the AR promoter reporter, whereas androgen-mediated expression of the AR promoter activity was markedly suppressed by RC, MM, and RD after 1 h treatments, and further declined in cells following prolonged treatments, suggesting that these chemicals transcriptionally inhibited AR expression. The corresponding induction of endogenous AR protein by androgen was also profoundly inhibited by RC, MM, and RD in LNCaP cells (), suggesting that RC, MM, and RD were able to inhibit AR expression at both mRNA and protein levels.
Figure 2. Analysis of AR expression in LNCaP after treated with RC, MM, or RD. (A) Quantitative PCR analysis of AR mRNA levels. ***p < 0.001 compared with the Mib+ control group. (B) AR promoter activity analysis by relative luciferase activity assays after treatment with the chemicals for 1 h and 24 h respectively. *p < 0.05, **p < 0.005, and ***p < 0.001 compared with the control group. (C) Western blotting analysis of AR.
We further examined the impact of bisbibenzyls on AR transcriptional activity by monitoring the activity of the promoter of prostate-specific antigen (PSA), a well-known AR target gene. As shown in , liganded AR-mediated stimulation of the PSA promoter was dramatically repressed by RC, MM, and RD in LNCaP cells. Inhibitory effect of bisbibenzyls on the transactivation of the AR was also evaluated in PC3 cell line, which lacks endogenous AR. The PSA promoter activity in cells transfected with the AR expression plasmid was markedly enhanced in response to Mib, whereas the Mib-mediated stimulation was significantly abolished in cells exposed to RC, MM, and RD (). To specifically verify the effect of bisbibenzyls on AR transcriptional activity, we examined luciferase activity of a reporter containing three tandem repeats of ARE of the hk2 gene, another AR target gene. As expected, exposure of LNCaP cells to RC, MM, and RD led to elimination of the reporter transcription in the presence of Mib (). Similar results were observed in PC3 cells (). ELISA assay revealed that Mib-stimulated endogenous PSA protein in cell culture media was significantly lower in cells exposed to RC, MM, and RD when compared with Mib treatment alone ().
Figure 3. The impact of RC, MM, and RD on AR transcriptional activity. Endogenous (A) and exogenous (B) AR transcriptional activities were analyzed by detecting the activity of the promoter of PSA. ***p < 0.001 compared with the control group. Endogenous (C) and exogenous (D) AR transcriptional activities were analyzed by detecting the activity of ARE. *p < 0.05 and ***p < 0.001 compared with the control group. (E) ELISA analysis of PSA in LNCaP cell media after treated with RC, MM, or RD. ***p < 0.001 compared with the control group. (F) Representative immunofluorescence staining of AR (Red) in LNCaP cells with/without compounds treatment. Nuclei were counterstained with DAPI (blue). (G) Cytoplasmic and nucleoplasmic levels of AR were detected by western blot after treated with RC, MM, or RD. (H) Associations of AR and SRC-1 were determined by coimmunoprecipitation using anti-AR.
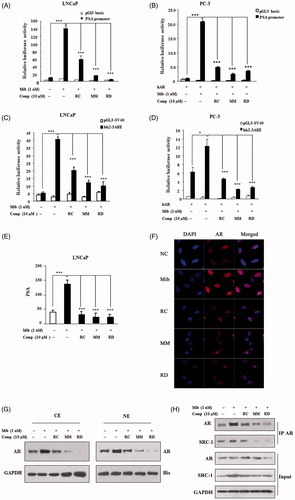
We next investigated if suppression of AR transactivation by bisbibenzyls resulted from their influence on the androgen-induced translocation of AR to the nucleus, which is believed to be critical for AR activity. As predicted, immunofluorescence staining revealed that Mib treatment predominantly stimulated the AR translocation from cytosol to the nucleus, leading to strong fluorescent intensity in the nucleus. However, the chemical treatments resulted in much less fluorescence in the nucleus, due to significantly impaired translocation of AR into nucleus in the presence of Mib (). This observation was further validated by western blotting as shown in , treatment of cells with RC, MM, and RD significantly reduced androgen-induced AR nuclear compartmentalization. Considering coactivators such as steroid receptor coactivator-1 (SRC-1) which potentiate AR-mediated transcriptional activity by conferring recruitment of CBP/p300 (Heinlein & Chang, Citation2002), we examined SRC-1/AR interactions using co-immunoprecipitation. As shown in , the expressions of SRC-1 in either chemical-treated or untreated cells almost remained unchangeable; however, after AR immunoprecipitation, SRC-1 levels were evident in the immunoprecipitated complexes of androgen-treated cells, whereas a decline in the level of SRC-1 protein in AR immuonprecipitates was noticeably observed in cells treated with RC, MM, and RD. Thus, downregulation of AR expression by bisbibenzyls resulted in the abolishment of AR activity in the nucleus because of the reduced AR nuclear localization and its decreased interaction with SRC-1 co-activator.
Bisbibenzyls impair proteasome activity
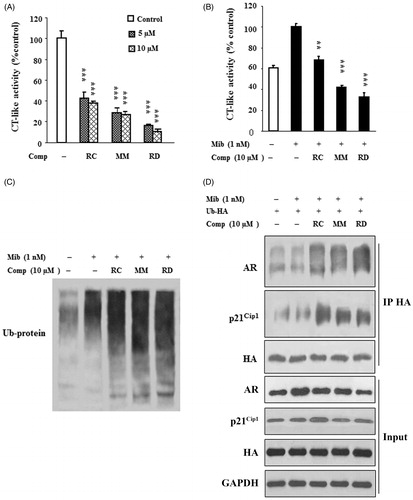
The ubiquitin/proteasome pathway is one of the mechanisms responsible for the regulation of AR expression and activity (Jaworski, Citation2006); therefore, we wanted to test whether proteasomal activity is involved in bisbibenzyl-mediated suppression of AR in the presence of androgen. First, proteasome activity assays with purified proteasome in the presence of bisbibenzyls were performed. The results in reveal that RC, MM, and RD significantly inhibited the proteasome activity as indicated by reduced levels of the CT-like activity of purified 20S proteasome, and the inhibitions were 57.5, 71.9, and 83.7% for RC, MM, and RD at 5 μM, respectively. Driven by the observations, we then tested if these bisbibenzyls can inhibit the 26S proteasome activity in LNCaP cell extracts. As shown in , CT-like activity was enhanced in cells treated with Mib, whereas RC, MM, and RD treatments inhibited the Mib-induced proteasomal CT-like activity in cell extracts, by 38.1, 58.2, and 67.2% at 10 μM, respectively. The concentrations of RC, MM, and RD required for the inhibition of the proteasome activity in cell extracts were somehow higher than those with purified 20S proteasome (), suggesting that decay of bisbibenzyls by cellular metabolism might occur in cells, consistent with the notion of the previous observations on EGCG (Nam et al., Citation2001). As the inhibition of the proteasome activity leads to the accumulation of polyubiquitinated proteins, our results displayed that the increases in polyubiquitinated-protein levels were noted in cells exposed to RC, MM, and RD in the presence of Mib (), supporting the observed inhibitory effect of bisbibenzyls on the proteasome activity. We further explored whether AR is polyubiquitinated in bisbibenzyl-treated cells after transfected with a HA-ubiquitin expression plasmid. The results presented in demonstrate that the AR proteins were decreased; however, the levels of polyubiquitinated-AR were noticeably increased in HA-precipitated immunocomplexes from cells treated with chemicals in the presence of Mib, suggesting that the increased polyubiquitinated-AR might be degraded in different pathways other than the ubiquitin/proteasome pathway. On the contrary, treatments with RC, MM, and RD increased the levels of ubiquitinated-p21Cip1 protein, while the total p21Cip1 protein levels were moderately increased in the chemical-treated cells. Nonetheless, the regulation of p21Cip1 protein by bisbiphenzyls remains to be investigated in the near future. Hence, the data clearly indicated that RC, MM, and RD inhibited proteasome activity, leading to the accumulation of polyubiquitinated proteins including AR in LNCaP cells.
Figure 4. RC, MM, and RD repress proteasome activity. (A) The compounds were used to directly act on purified 20S proteasome, and proteasome activities were detected. Columns, mean (n = 3); bars, SD; ***p < 0.001 compared with the control group (DMSO treatment). (B) LNCaP cells were treated with the compounds for 24 h, and cell lysates were performed to detect proteasome activities. Columns, mean (n = 3); bars, SD; **p < 0.005 and ***p < 0.001 compared with the control group. (C) Western blotting analysis of ubiquitinated protein in LNCaP cells after treated with RC, MM, or RD. (D) Polyubiquitinated-AR and polyubiquitinated-p21Cip1 were detected by coimmunoprecipitation using anti-HA after transfection of a HA-ubiquitin expression plasmid in LNCaP cells.
Bisbibenzyls activate autophagy
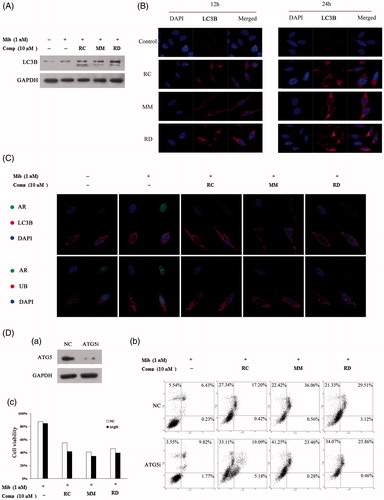
Given studies demonstrating that autophagy, another major intracellular protein degradation system, is induced in response to proteasome inhibition under certain situations (Ding et al., Citation2007), we, therefore, hypothesized that bisbibenzyl-mediated proteasome inhibition simultaneously activates autophagy in PCa cells, which in turn is involved in degrading the polyubiquitinated-AR. In our study, induction of LC3B expression and conversion was found in LNCaP cells treated with the compounds (). Moreover, the formation of LC3 puncta, representing association of LC3B-II with autophagosomes, was significantly induced in cells exposed to the compounds (). Thus, these results demonstrated the ability of autophagy induction by these agents. To investigate whether autophagy is involved in degrading the ubiquitinated AR, we performed immunofluorescence co-location analysis on AR and LC3 or Ub. As demonstrated in , prior to pretreatment with the chemicals, AR showed nuclear accumulation with intense nucleoplasmic fluorescence in response to Mib treatment. After treatment with the compounds, an altered localization of AR was found and cells showed a co-localization of AR and LC3 or Ub, suggesting that the activation of autophagy by RC, MM, and RD might be involved in the process of ubiquitinated AR degradation. Moreover, a knockdown experiment was performed using siRNA targeting Atg5, an important gene contributing to autophagic vesicle formation, to investigate the effect of autophagy induced by these chemicals on cell survival. The results indicated that depletion of autophagy by knocking down Atg5 only slightly decreased cell survival. Blockage of bisbibenzyl-activated autophagy had no significant effect on MM-mediated apoptosis, but facilitated RC- and RD-induced apoptosis, suggesting that the effects of activation of autophagy on apoptosis are dependent on types of the chemicals used ().
Figure 5. Induction of autophagy by RC, MM, or RD in LNCaP cells. (A) Western blot analysis of LC3 in compound-treated LNCaP cells. (B) Representative images of the punctuated LC3 in LNCaP cells treated with RC, MM, or RD. (C) Representative immunofluorescence staining of AR (green) and LC3 or Ub (red) in compound-treated LNCaP cells. Nuclei were counterstained with DAPI (blue). (D) (a) Western blotting was performed to confirm ATG5 knockdown; (b) percentage of apoptotic cells was analyzed by flow cytometry after ATG5 knockdown; (c) cell viability assays after depletion of autophagy using Atg5-targeting siRNA in LNCaP cells prior to treatment with the chemicals.
Discussion
AR plays an important role in the proliferation and survival of PCa cells, whereas down-regulation of AR can trigger proapoptotic events. This study reveals a novel finding that RC, MM, and RD, three naturally occurring macrocyclic bisbibenzyl agents, effectively down-regulated AR expression and function, and caused both G1 arrest and induction of cell death via apoptosis. Treatments of cells with RC, MM, and RD resulted in induction of p21Cip1 and E2F-1, which caused cell cycle arrest. The compounds also induced an increase in Bax, and a decrease in Bcl-2, thus promoting apoptosis. E2F-1 is a well-documented regulatory protein required for G1-S transition and DNA synthesis. Recent findings demonstrated that E2F-1 acts as an oncogene or exerts a tumor-promoting role at low levels, but induces apoptosis when its expression is high or upregulated. Overexpression of E2F-1 is associated with AR down-regulation in CRPC (Davis et al., Citation2006; Johnson & Degregori, Citation2006). We now observed that RC, MM, and RD moderately enhanced E2F-1 in the presence of Mib in LNCaP cells. Given the observed basal level of E2F-1 in LNCaP cells, we reasoned that induction of E2F-1 together with downregulation of AR by bisbibenzyls exerted proapoptotic effect. However, thymoquinone, a compound from herb Nigella sativa, prevents cell growth by blocking expression of the AR, E2F-1, and E2F-1-regulated cell-cycle-associated proteins in PCa cells (Kaseb et al., Citation2007). Therefore, the role of E2F-1 in bisbibenzyl-mediated apoptosis might not be simply dependent on its expression levels, therefore, requires to be further elucidated. Our results displayed that inhibition of RC, MM, and RD on AR expression led to significant decreases in the AR nuclear level and association of AR/SRC-1 in the nucleus with consequent reduction of PSA protein and AR-regulated promoter activities, suggesting the importance of downregulation of AR expression and activities in their antiproliferative and proapoptotic effects.
Considering the multiple mechanisms involved in the modulation of AR expression and functions, the ubiquitin/proteasome system can be regarded as a complicated mechanism in this regulatory process. The AR protein levels can be controlled by ubiquitin/proteasome-dependent degradation in the absence or presence of androgen. The inhibition of proteasome results in an increase in the polyubiquitylated AR forms (Jaworski, Citation2006; Sheflin et al., Citation2000). MG132, a well-characterized proteasome inhibitor, is able to decrease AR protein expression after 24 h treatment in LNCaP cells, which correlates with the observation that the mRNA level of AR is also reduced with a long-term treatment. This may be explained as MG132 has effects that entail suppression of AR mRNA expression yet accumulation of polyubiquitinated AR (Jaworski, Citation2006). Consistent with this finding, we also observed that bisbibenzyl-mediated inhibitory effects on AR were associated with blocking of proteasome activity as evidenced by the decreased CT-like activity and accumulation of polyubiquitinated proteins including AR. In addition, the components of the ubiquitin/proteasome system and proteasome activity are implicated to regulate AR transcriptional activity (Jaworski, Citation2006; Lin et al., Citation2002). Lin et al. (Citation2002) indicate that MG132 can interrupt the interaction between AR and ARA70 or TIF2, leading to abnormal AR action. Our finding revealed that RC, MM, and RD impaired the association of AR and SRC-1, and decreased the AR activity. In accordance with the concept that proteasome inhibitors have potential to regulate AR expression and functions (Reddy et al., Citation2006), our results indicated that bisbibenzyl treatments caused downregulation of AR transcription, suppression of the transcriptional activity of AR via decreased nuclear translocation of AR, and inhibition of interaction between AR and its co-regulators. However, further studies are required to address whether and how bisbibenzyls can disrupt the interactions of AR/SRC-1 or other coregulators in PCa cells.
Most recently, genetic depletion of AR in LNCaP or CWRrv1 cells, or pharmacologic blockage of AR with the anti-androgen bicalutamide, led to the induction of autophagy (Boutin et al., Citation2013; Jiang et al., Citation2012), supporting our observations that suppression of AR by bisbibenzyls promoted autophagy in LNCaP cells. Our data demonstrated that bisbibenzyl treatments predominantly caused co-localization of AR/LC-3B, and AR/Ub in the cytosol, suggesting that activation of autophagy by the chemicals is involved in ubiquitinated AR degradation. Further studies will be required to address the mechanisms by which bisbibenzyl induce autophagy for degradation of the AR. We further inactivated autophagy by autophagy inhibitor to examine whether autophagy induced simultaneously by these bisbibenzyls is a sign for cytoprotective response that is frequently observed in tumor cells exposed to chemotherapy or radiation (Kondo et al., Citation2005; Zhu et al., Citation2010). As shown, suppression of autophagy had no significant effect on MM-mediated apoptosis, but facilitated RC- and RD-induced apoptosis, suggesting that activation of autophagy is dependent on types of the chemicals used. We have demonstrated that MM has ability to trigger autophagic cell death in addition to the induction of cellular apoptosis in androgen-independent PC3 (Jiang et al., Citation2013). Therefore, the role of autophagy induced by bisbibenzyls may also depend on cell-types with particular cellular contexts. For example, because PC3 cells are p53-null, we would want to test the possibility whether MM-induced p53 may play a critical role in the regulation of autophagy in LNCaP cells. Tumor suppressor p53 is activated in response to various stress signals, and activated p53 is important for cell-cycle arrest or for apoptosis induction through regulation of its target genes such as p21Cip1. In agreement with previous findings, we found that increases in the levels of p53 and p21Cip1 with RC, MM, and RD () might, in part, contribute to the observed cell-cycle arrest and apoptosis. Recent findings demonstrate that p53 also participates in proteasome-dependent modulation of AR activity and induction of autophagy, and the inhibitory effects of proteasome inhibitors on AR are dependent on p53 status in PCa (Guseva et al., Citation2012). However, studies are required in future to establish the role of p53, in addition to its regulatory functions in cell cycle and apoptosis, in bisbibenzyl-mediated suppression of proteasome and AR.
In summary, our findings demonstrate that in addition to blocking cell division, bisbibenzyls impair AR signaling, suggesting the therapeutic potential of bisbibenzyls in the treatment of PCa.
Acknowledgements
We would like to thank Prof. Chengjiang Gao (Department of immunology, Shandong University School of Medicine) for the generous gift of HA-Ub expression plasmid.
Declaration of interest
This work was supported by the National Natural Science Foundation of China (30772594, 30973551, and 30925038), and Shandong Scientific Technology Program (2012GSF11909).
References
- Adams J. (2004). The development of proteasome inhibitors as anticancer drugs. Cancer Cell 5:417–21
- Bluemn EG, Nelson PS. (2012). The androgen/androgen receptor axis in prostate cancer. Curr Opin Oncol 24:251–7
- Boutin B, Tajeddine N, Vandersmissen P, et al. (2013). Androgen deprivation and androgen receptor competition by bicalutamide induce autophagy of hormone-resistant prostate cancer cells and confer resistance to apoptosis. The Prostate 73:1090–102
- Chen D, Cui Q-C, Yang H, et al. (2007). Clioquinol, a therapeutic agent for Alzheimer's disease, has proteasome-inhibitory, androgen receptor-suppressing, apoptosis-inducing, and antitumor activities in human prostate cancer cells and xenografts. Cancer Res 67:1636–44
- Chymkowitch P, Le-May N, Charneau P, et al. (2011). The phosphorylation of the androgen receptor by TFIIH directs the ubiquitin/proteasome process. EMBO J 30:468–79
- Davis JN, Wojno KJ, Daignault S, et al. (2006). Elevated E2F1 inhibits transcription of the androgen receptor in metastatic hormone-resistant prostate cancer. Cancer Res 66:11897–906
- Ding WX, Ni HM, Gao W, et al. (2007). Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 171:513–24
- Feldman BJ, Feldman D. (2011). The development of androgen-independent prostate cancer. Nat Rev Cancer 1:34–45
- Guseva NV, Rokhlin OW, Glover RA, Cohen MB. (2012). P53 and the proteasome regulate androgen receptor activity. Cancer Biol Ther 13:553–8
- Heinlein CA, Chang C. (2002). Androgen receptor (AR) coregulators: An overview. Endocr Rev 23:175–200
- Huang H, Zegarra-Moro OL, Benson D, Tindall DJ. (2004). Androgens repress Bcl-2 expression via activation of the retinoblastoma (RB) protein in prostate cancer cells. Oncogene 23:2161–76
- Jaworski T. (2006). Degradation and beyond: Control of androgen receptor activity by the proteasome system. Cell Mol Biol Lett 11:109–31
- Jiang H, Sun J, Xu Q, et al. (2013). Marchantin M: A novel inhibitor of proteasome induces autophagic cell death in prostate cancer cells. Cell Death Dis 4:e761
- Jiang Q, Yeh S, Wang X, et al. (2012). Targeting androgen receptor leads to suppression of prostate cancer via induction of autophagy. J Urol 188:1361–8
- Jiang SW, Eberhardt NL. (1995). A micro-scale method to isolate DNA-binding proteins suitable for quantitative comparison of expression levels from transfected cells. Nucleic Acids Res 23:3607–8
- Johnson DG, Degregori J. (2006). Putting the oncogenic and tumor suppressive activities of E2F into context. Curr Mol Med 6:731–8
- Kar G, Keskin O, Fraternali F, Gursoy A. (2013). Emerging role of the ubiquitin-proteasome system as drug targets. Curr Pharm Des 19:3175–89
- Kaseb AO, Chinnakannu K, Chen D, et al. (2007). Androgen receptor and E2F-1 targeted thymoquinone therapy for hormone-refractory prostate cancer. Cancer Res 67:7782–8
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. (2005). The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 5:726–34
- Lin H, Altuwaijri S, Lin W, et al. (2002). Proteasome activity is required for androgen receptor transcriptional activity via regulation of androgen receptor nuclear translocation and interaction with coregulators in prostate cancer cells. J Biol Chem 277:36570–6
- Loidl G, Groll M, Musiol HJ, et al. (1999). Bivalency as a principle for proteasome inhibition. Proc Natl Acad Sci USA 96:5418–22
- Lu Z, Fan P, Ji M, Lou H. (2006). Terpenoids and bisbibenzyls from Chinese liverworts Conocephalum conicum and Dumortiera hirsuta. J Asian Nat Prod Res 8:187–92
- Micel LN, Tentler JJ, Smith PG, Eckhardt GS. (2013). Role of ubiquitin ligases and the proteasome in oncogenesis: Novel targets for anticancer therapies. J Clin Oncol 31:1231–8
- Mori A, Lehmann S, O'Kelly J, et al. (2006). Capsaicin, a component of red peppers, inhibits the growth of androgen-independent, p53 mutant prostate cancer cells. Cancer Res 66:3222–9
- Nam S, Smith D, Dou Q. (2001). Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. J Biol Chem 276:13322–30
- Qu J, Xie C, Guo H, et al. (2007). Antifungal dibenzofuran bis(bibenzyl)s from the liverwort Asterella angusta. Phytochemistry 68:1767–74
- Reddy G, Barrack E, Dou Q, et al. (2006). Regulatory processes affecting androgen receptor expression, stability, and function: Potential targets to treat hormone-refractory prostate cancer. J Cell Biochem 98:1408–23
- Seruga B, Ocana A, Tannock IF. (2011). Drug resistance in metastatic castration-resistant prostate cancer. Nat Rev Clin Oncol 8:12–23
- Sheflin L, Keegan B, Zhang W, Spaulding S. (2000). Inhibiting proteasomes in human HepG2 and LNCaP cells increases endogenous androgen receptor levels. Biochem Biophys Res Commun 276:144–50
- Shi Y, Liao Y, Qu X, et al. (2008). Marchantin C, a macrocyclic bisbibenzyl, induces apoptosis of human glioma A172 cells. Cancer Lett 262:173–82
- Shi Y, Zhu C, Yuan H, et al. (2009). Marchantin C, a novel microtubule inhibitor from liverwort with anti-tumor activity both in vivo and in vitro. Cancer Lett 276:160–70
- Xu A, Hu Z, Qu J, et al. (2010). Cyclic bisbibenzyls induce growth arrest and apoptosis of human prostate cancer PC3 cells. Acta Pharmacol Sin 31:609–15
- Xue X, Qu X, Gao Z, et al. (2012). Riccardin D, a novel macrocyclic bisbibenzyl, induces apoptosis of human leukemia cells by targeting DNA topoisomerase II. Invest New Drugs 30:212–22
- Yang H, Shi G, Dou Q. (2007). The tumor proteasome is a primary target for the natural anticancer compound Withaferin A isolated from “Indian winter cherry”. Mol Pharmacol 71:426–37
- Yeap B, Voon D, Vivian J, et al. (2002). Novel binding of HuR and poly(C)-binding protein to a conserved UC-rich motif within the 3′-untranslated region of the androgen receptor messenger RNA. J Biol Chem 277:27183–92
- Yuan H, Kong F, Wang X, et al. (2008). Inhibitory effect of acetyl-11-keto-beta-boswellic acid on androgen receptor by interference of Sp1 binding activity in prostate cancer cells. Biochem Pharmacol 75:2112–21
- Zhu K, Dunner K-Jr, McConkey D. (2010). Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 29:451–62
- Zhu M, Kyprianou N. (2008). Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr Relat Cancer 15:841–9