
Abstract
Context: Euphorbia hirta L. (Euphorbiaceae) has been used as a folk remedy in Southeast Asia for the treatment of various ailments.
Objective: The current study evaluates the cytotoxicity, cell-cycle arrest, and apoptotic induction by E. hirta in MCF-7 breast cancer cells.
Materials and methods: Cytotoxic activity of methanol extract of whole part of E. hirta was determined by the MTT assay at various concentrations ranging from 1.96 to 250.00 µg/mL in MCF-7 cells. Cell morphology was assessed by light and fluorescence microscopy. Apoptosis and cell-cycle distribution were determined by annexin V staining and flow cytometry. DNA fragmentation, caspase activity, and reactive oxygen species (ROS) assays were performed using the commercially available kits. To identify the cytotoxic fraction, E. hirta extract was subjected to bioassay-guided fractionation.
Results: Euphorbia hirta exhibited significant inhibition of the survival of MCF-7 cells and the half inhibitory concentration (IC50) values was 25.26 µg/mL at 24 h. Microscopic studies showed that E. hirta-treated cells exhibited marked morphological features characteristic of apoptosis. Euphorbia hirta extract also had an ignorable influence on the LDH leakage and generating intracellular ROS. The flow cytometry study confirmed that E. hirta extract induced apoptosis in MCF-7 cells. Euphorbia hirta also resulted in DNA fragmentation in MCF-7 cells. Moreover, E. hirta treatment resulted in the accumulation of cells at the S and G2/M phases as well as apoptosis. The caspase activity study revealed that E. hirta extract induced apoptosis through the caspase-3-independent pathway by the activation of caspase-2, 6, 8, and 9. Euphorbia hirta hexane fraction, namely HFsub4 fraction, demonstrated highest activity among all the fractions tested with an IC50 value of 10.01 µg/mL at 24 h.
Discussion and conclusion: This study revealed that E. hirta induced apoptotic cell death and suggests that E. hirta could be used as an apoptosis-inducing anticancer agent for breast cancer treatment with further detailed studies.
Introduction
In the last decade, cancer research has made remarkable advancements in our basic understanding and approach to cancer research and cancer biology. Recent evidence indicates the vital roles of apoptosis in the development of therapeutic agents and treatment of cancer (Hassan et al., Citation2014). The goal of cancer therapy is to promote the death of cancer cells without causing too much damage to normal cells (Gerl & Vaux, Citation2005). Our knowledge of the mechanisms of apoptosis has enhanced our understanding of how some cancers originate and progress. It has also revealed that existing cancer therapies can work in two ways, by induction of apoptosis and by direct toxicity (Gerl & Vaux, Citation2005). Hence, the regulators of cell death and the cell-cycle checkpoint are the targets in the development of therapeutic agents and the treatment of cancer (Tyagi et al., Citation2002). Currently, the contribution of cell cycle checkpoint regulation to synergically enhance cell death is largely unknown. Therefore, both checkpoint abrogation and checkpoint arrest, as a means to enhance the cytotoxic effects of chemotherapy, require additional study (Tyagi et al., Citation2002). The current study was designed to develop a novel therapeutic agent from natural resources to exploit our knowledge of apoptotic mechanisms to promote apoptosis of cancer cells and limit the concurrent death of normal cells. The medicinal plants used in traditional medicine in developing countries contain a wide range of phytochemicals that can be used to treat cancer (Kraft, Citation2009). One such plant known to have curative value is Euphorbia hirta L. (Euphorbiaceae). As E. hirta is a commonly used herbal remedy that has been reported to possess various pharmacological activities, including anticancer activity, the aim of this study was to investigate the impact of E. hirta on cancer cell lines in vitro to ascertain if it possesses any potential chemotherapeutic activity. The cell lines used were the Vero (green African monkey kidney cells), MCF-7 (breast adenocarcinoma cells), HT-29 (colon adenocarcinoma cells), and Hela (cervical adenocarcinoma cells) cell line.
Euphorbia hirta is a small annual herb common in tropical countries. It is frequently seen occupying open waste spaces, banks of watercourses, grasslands, roadsides, and pathways (Mamun-Or-Rashid et al., Citation2013; Rajeh et al., Citation2010). Euphorbia hirta is a very popular herb amongst traditional medicine practitioners and is traditionally used for the treatment of various ailments, including intestinal parasites, diarrhea, peptic ulcers, heartburn, vomiting, amoebic dysentery, asthma, bronchitis, hay fever, laryngeal spasms, emphysema, coughs, colds, kidney stones, menstrual problems, sterility, and venereal diseases (Basma et al., Citation2011b). Furthermore, the plant is also applied to treat infections of the skin and mucous membranes, including warts, scabies, tinea, thrush, aphthae, fungal afflictions, measles, Guinea-worm, and as an antiseptic to treat wounds, sores and conjunctivitis. Euphorbia hirta is also employed as an analgesic to treat severe headaches, toothache, rheumatism, colic pains, and also pains during pregnancy (Kumar et al., Citation2010). It is used as an antidote and a pain relief for scorpion stings and snakebites. The use of the latex from E. hirta to facilitate the removal of thorns from the skin is common (Shih & Cherng, Citation2012). The sedative, anxiolytic, analgesic, antipyretic, and anti-inflammatory properties of E. hirta have been reported in the literature (Shih & Cherng, Citation2012). The leaf extract of E. hirta increased the urine output and electrolytes in rats (Johnson et al., Citation1999; Lanhers et al., Citation1991). Furthermore, studies revealed that E. hirta possesses galactogenic, anti-anaphylactic, antimicrobial, antioxidant, anticancer, antifeedant, antiplatelet aggregation, anti-inflammatory, aflatoxin inhibition, antifertility, anthelmintic, antiplasmodial, antiamoebic, antimalarial, and larvicidal activities against Plutella xylostella (Rajeh et al., Citation2010). Our latest research showed that the E. hirta methanol extract exhibited only mild toxic effects in mice in an oral acute toxicity study (Yuet Ping et al., Citation2013).
Materials and methods
Chemicals
DMEM, FBS, trypsin, penicillin, and streptomycin were purchased from Gibco (Invitrogen, Grand Island, NY). 3-(4,5-Dimethyl thiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), were purchased from Sigma-Aldrich (St. Louis, MO). Annexin V-FITC and PI double staining kit were purchased from BD (Waltham, MA). Doxorubicin hydrochloride (Sigma, St. Louis, MO) was the standard drugs used as positive control in this study.
Preparation of E. hirta methanol extract
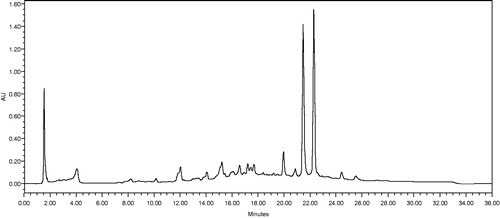
Euphorbia hirta was collected from various areas in Universiti Sains Malaysia, Penang, in January 2012 and was identified by Mr. Shanmugam Vellosamy at the Herbarium of School of Biological Sciences, Universiti Sains Malaysia, Pulau Pinang, Malaysia, where the voucher specimen was deposited (Number: USM/HERBARIUM/11215). Whole part of E. hirta was thoroughly rinsed with tap water and distilled water before being air-dried at room temperature for 7 d. Then, the plant samples were ground to fine powder and soaked (approximately 100 g of the dried plant material) in absolute methanol (400 mL) for 4 d with frequent agitation. The extract was filtered with Whatman paper No. 1 and the residue was then re-soaked with a fresh portion of methanol twice. The filtrate was then concentrated by rotary evaporator (BUCHI Rotary Evaporator R110, Buchi, Flawil, Switzerland) under reduced pressure. The yield of the extract was 17.1% based on dry weight. The dried residue of plant extract was resuspended in DMSO (Sigma, St. Louis, MO) for further biological assays. The HPLC chromatogram of E. hirta is shown in . The chromatographic analyses were performed on a Novapak C18 column (4.6 × 250 mm) at ambient temperature with mobile phase of methanol/water/formic acid (50:49:1, v/v) at a flow rate of 1 mL/min. To standardize the fingerprint, 10 batches of E. hirta samples were analyzed with the developed procedure. The peaks that existed in all ten batches of samples were assigned as “common peaks” for the E. hirta extract (). There are 18 “common peaks” in the fingerprint. Mok and Chau (Citation2006) recommended the authentication and quality control of herbal medicines using a pattern-based approach because of the chemical ingredients of most medicinal plants that have been studied; however, it is difficult to determine which compound is effective. The method developed for HPLC fingerprinting provided a quick analysis of the methanolic extract of E. hirta and serves as a reference in the identification of authentic E. hirta methanol extract for future work.
Figure 1. The HPLC “fingerprints” of the methanolic extracts of E. hirta showed major eighteen “common peaks” (higher concentration of components) at various retention times (min) at a wavelength of 254 nm.
Cell lines
Vero (green African monkey kidney cells), MCF-7 (breast adenocarcinoma cells), HT-29 (colon adenocarcinoma cells), and Hela (cervical adenocarcinoma cells) were obtained from the American Type Culture Collection (ATCC, Manassas, VA). All cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS), penicillin–streptomycin 1% (v/v) (Gibco, Grand Island, NY). The cells were cultured in 5% CO2 incubator at 37 °C in a humidified atmosphere.
MTT cytotoxicity assay
Cytotoxicity activity of E. hirta was determined by using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) tetrazolium reduction assay (Mosmann, Citation1983). Four cells lines were selected to be investigated throughout this study; the cancerous MCF-7, HT-29, Hela cell lines, and the control serving non-cancerous Vero cell lines. The dried residue of plant extract was resuspended in DMSO (Sigma, St. Louis, MO) for further biological assays to obtain a stock solution of 10.00 mg/mL. The substock solution was prepared by diluting the stock solution by using culture medium. The final concentration of DMSO in all the experiments did not exceed 0.5% (v/v). Cells were seeded at a density of 1 × 104 cells per well in 96-well plates overnight and then treated with different concentrations of E. hirta (250.00, 125.00, 62.50, 31.25, 15.63, 7.81, 3.91, and 1.96 μg/mL). After 24, 48, and 72 h treatment, 20 μL of MTT solution (5 mg/mL in PBS) were added to each well and the cells were further incubated at 37 °C for 4 h. Subsequently, the medium was aspirated and 100 μL of DMSO was added to solubilize the water-insoluble purple formazan crystals. The plates were shaken and the optical density was measured at 570 nm using a microplate reader (Thermo Scientific Multiskan Spectrum, Thermo Fisher Scientific, Inc., Waltham, MA). The IC50 values of E. hirta extract on various cell lines were determined using Graph Pad Prism version 3.00 (GraphPad, San Diego, CA) and used in subsequent experiments. Doxorubicin hydrochloride was used as a positive control. Results were expressed as mean ± standard deviation (SD).
Morphological detection of apoptosis using light microscopy
Light microscopic examination of the cells was performed to observe the morphological changes after treatment. MCF-7 cells were grown in 6 well plates and treated with E. hirta at a concentration of 25.00 μg/mL for 24, 48, and 72 h. The morphological changes were observed under inverted light microscope (Olympus, Hamburg, Germany).
Morphological detection of apoptosis using acridine orange/propidium iodide (AO/PI) staining
The mode of cell death induced by extract was examined using AO/PI staining. Briefly, treated cells were harvested from the culture flask. After 24, 48, and 72 h, the cell suspension was mixed with 10 μL of AO (1 mg/mL) and 10 μL of PI (1 mg/mL) (Mishell et al., Citation1980). The mixture (20 μL) was aliquoted onto a slide and covered with a cover slip and viewed under fluorescence microscope. The mode of cell death was then determined. Viable, apoptotic, and necrotic cells were identified using fluorescence microscopic observation (Carl Zeiss, Germany) on the basis of distinct morphological changes, including membrane blebbing, nuclear and cytosolic condensation and nuclear fragmentation. A minimum of 200 cells were counted per slide. Untreated MCF-7 cells served as the control.
Lactate dehydrogenase leakage assay
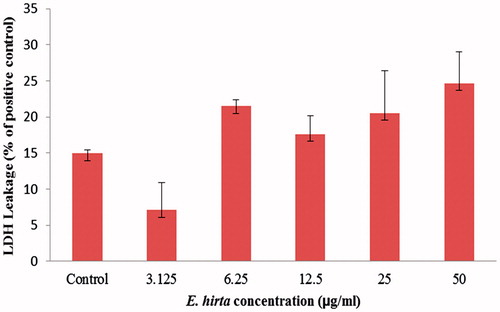
Lactate dehydrogenase (LDH) cytotoxicity assay kit (Cayman Chemical Co., Ann Arbor, MI) was employed to assess the cell membrane integrity. The assay was based on the measurement of activity of LDH released from the damaged cells into the medium (Kim et al., Citation2008). Cells were plated in the 96-well plates (1 × 104 cells per well) and incubated for 24 h. Euphorbia hirta extract was introduced separately to the cells with different concentrations (50, 25, 12.5, 6.25, 3.125, and 1.5625 μg/mL) and incubated for another 24 h. The positive control was prepared by adding 1% Triton of lysis solution to the control cells at 45 min prior to the centrifugation. The cells were centrifuged at 1200 rpm for 5 min and 100 µL of the supernatant of each well was transferred to a new flat bottom 96-well culture plate. The absorbance at 490 nm was recorded on a microplate reader (Thermo Scientific Multiskan Spectrum, Thermo Fisher Scientific, Inc., Waltham, MA). The LDH leakage (% of positive control) is expressed as the percentage of (ODtest−ODblank)/(ODpositive−ODblank), where ODtest is the optical density of the control cells or cells exposed to E. hirta extract, ODpositive is the optical density of the positive control cells and ODblank is the optical density of the wells without cells.
Reactive Oxygen Species Assay (ROS assay)
The oxidant-sensitive dye DCFH-DA was used for ROS detection (OxiSelect Intracellular ROS Assay Kit, Cell Biolabs, San Diego, CA). Cells were plated in the 96-well plates (2 × 104 cells per well) and incubated for 24 h. After 24 h, the medium was removed and the wells were washed with 200 µL phosphate buffer saline (PBS). About 100 μL of 1 × DCFH-DA/media solutions were added to each well and incubate at 37 °C for 30 min in the dark. The solution were then removed and washed with PBS several times. Euphorbia hirta extract was introduced to the DCFH-DA-loaded cells with different concentrations (200, 100, 50, 25, 12.5, 6.25, 3.125, and 1.5625 μg/mL). The positive controls were prepared by culturing the normal cells with culture medium containing 200 mM H2O2 at 1 h prior to the addition of DCFH-DA probe. The fluorescence generated by the DCFH oxidation was measured using fluorescence microplate reader (SpectraFluor, TECAN, Sunrise, Austria) with excitation and emission wavelengths of 480 and 530 nm, respectively. Data points were taken every 5 min for 30 min and the data were exported to Excel spreadsheet software for analysis. The ROS level is expressed as the ratio of (Ftest−Fblank)/(Fcontrol−Fblank), where Ftest is the fluorescence intensity of the cells exposed to E. hirta extract or the positive control, Fcontrol is the fluorescence intensity of the control cells, and Fblank is the fluorescence intensity of the wells without cells.
Annexin-V/PI dual staining assay
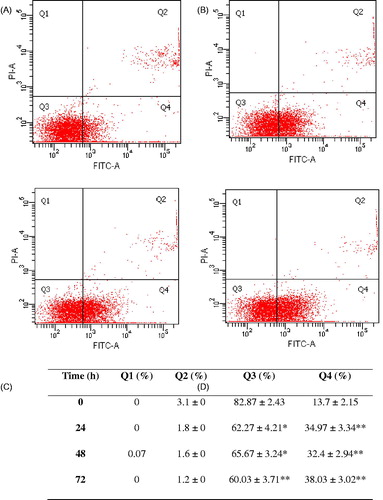
A quantitative assessment of apoptosis was performed using fluorescein isothiocyanate (FITC) Annexin V Apoptosis Detection Kit I (BD Biosciences, Waltham, MA). Briefly, cells were cultured (1.5 × 105 cells) overnight in 25 cm2 cell culture flasks prior to the treatment with E. hirta extract. Cells were then treated with IC50 value of the extract (25.00 μg/mL) for 24, 48, and 72 h. The positive control was prepared by culturing the control cells in medium containing 200 mM H2O2 for 30 min. Cells were collected, washed twice with cold PBS, and re-suspended in binding buffer (1 × 106 cells/mL). After 100 µL of cells was transferred to a tube, 5 µL of FITC-conjugated Annexin V (Annexin V-FITC) and 5 µL of propidium iodide (PI) were added followed by incubation for 15 min at room temperature in the dark. The stained cells were diluted by the binding buffer and immediately analyzed by the flow cytometer (Becton Dickinson FACSCanto II (BD FACSCanto II, BD Biosciences, Waltham, MA)). Data from 10 000 cells were collected in each data file. Four different populations of cells are easily distinguished: those that are unlabelled (viable cells), those that have bound Annexin V-FITC only (early apoptotic), those that have been stained with PI (necrotic), and those that have both bound Annexin V-FITC and been labelled with PI (late apoptotic/necrotic cells). The fluorescence distribution was displayed as a two-color dot plot analysis, and the fluorescent cells % in each quadrant was determined.
DNA fragmentation assay
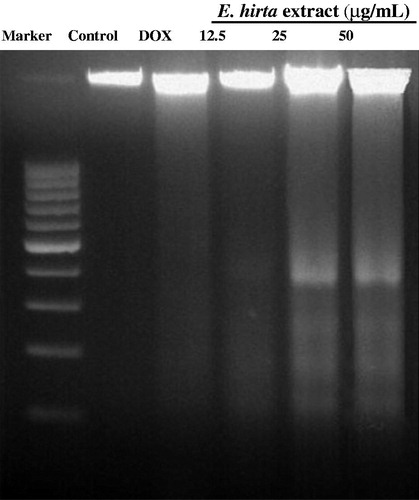
The detection of DNA ladder in agarose gel electrophoresis is used as a biochemical marker for the measurement of apoptosis. It is used to determine the presence of internucleosomal DNA cleavage. Briefly, MCF-7 cells were cultured overnight in 25 cm2 cell culture flasks prior to the treatment with E. hirta extract. Cells were then treated with E. hirta extract for 24 h. MCF-7 cells were collected, washed with PBS and purified using a DNA purification kit, DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA) according to the procedure of the manufacturer. The DNA was resolved by electrophoresis on 1.8% agarose gel at 80–100 V. The gel was stained with ethidium bromide, and DNA was visualized by a UV transilluminator.
Cell-cycle analysis
Each phase of the cell cycle was evaluated using DNA flow cytometry analysis. MCF-7 cells were cultured in 6-well plates overnight and then treated with different concentrations of E. hirta extract (12.5, 25.0, and 50.0 μg/mL). Doxorubicin (0.40 µg/mL) was used as a positive control. The Cycle TEST PLUS DNA Reagent Kit (Becton Dickinson Pharmingen, San Jose, CA) was used to determine the cell-cycle phase distributions. Nuclei were isolated, stained with PI, and analyzed on FACScan flow cytometer (Becton Dickinson, San Jose, CA) according to the instructions of the manufacturer. The percentages of cells in each cell-cycle phase were analyzed using Modfit software (ModFit, Topsham, ME) (Dobashi et al., Citation2003).
Detection of caspase activity by colorimetric assay
For evaluation of caspase activity, MCF-7 cells were cultured and treated with or without 25 µg/mL E. hirta extract. Cells were harvested and tested for caspase activity using the ApoTarget Caspase Colorimetric Protease Assay Sampler Kit (Invitrogen, Carlsbad, CA) according to the protocol of the manufacturer. Briefly, the cells were resuspended in 50 µL of chilled cell lysis buffer and incubated on ice for 10 min. The lysates were centrifuged at 10 000 g for 1 min. Protein concentration were determined using Bradford’s method. The protein (200 µg) was added to 50 µL 2 × reaction buffer containing 200 mM substrate VDVAD-pNA (caspase-2), DEVD-pNA (caspase-3), VEID (caspase-6), IETD-pNA (caspase-8), and LEHD-pNA (caspase-9), and incubated at 37 °C for 2 h in a 96-well plate. The absorbance of pNA was read at 405 nm in a microplate reader (Thermo Scientific Multiskan Spectrum, Thermo Fisher Scientific, Inc., Waltham, MA). Comparison of the absorbance of pNA from an apoptotic sample with an uninduced control allows the determination of the relative caspase activity (expressed as % of control).
Chromatographic fractionation of extracts of E. hirta for bioassay
The E. hirta extract (1.0 g) was dissolved in methanol:water (90:10, v/v) before was further partitioned in hexane:methanol:water (100:90:10, v/v/v) and yielded 0.085 g of hexane fraction (HF). Subsequently the aqueous layer formed was further partitioned in ethyl acetate (100 mL) to yield about 0.299 g of ethyl acetate fraction (EAF). Consequently, the aqueous layer formed was further partitioned in butanol (100 mL) and yielded 0.092 g of butanol fraction (BF). The remaining aqueous (AF) layer was collected as water fraction and yielded 0.452 g. The entire fractions were evaporated to dryness in a rotary evaporator (BUCHI Rotary Evaporator R110, Buchi, Switzerland). Each fraction was tested for their cytotoxic activity using MTT assay on MCF-7 cell line (Mosmann, Citation1983). The cytotoxic active HF, EAF, and BF were then further purified by using Medium Pressure Liquid Chromatography (MPLC) (Combi Flash Companion (Isco), Lincoln, NE). The column [4g RediSep flash column (CV 4.8–18 mL/min flow range), max pressure 600 psi, Combi Flash Companion (Isco), Lincoln, NE], was packed with silica gel (Merck, Durham, NC, silica gel 60). The column, loaded with the active HF, EAF and BF fraction, was eluted with hexane–acetone with gradient concentrations [10–100 v/v (%)]. Altogether, 22, 20, and 91 MPLC-sub fractions of 16.8 (HF), 16.8 (EAF), and 25.0 (BF) mL were collected, respectively, throughout the separation and elution procedure. Peaks were detected by UV at 254 nm by a DAD detector. All sub-fractions of the same compound were combined, and evaporated. HF and EAF fractions afforded eight sub-fractions each while BF fraction yielded 13 sub-fractions. All sub-fractions were evaporated to dryness under reduced pressure using rotary evaporator (BUCHI Rotary Evaporator R110, Buchi, Switzerland) before being subjected to the MTT assay with the MCF-7 cell line.
Results and discussion
MTT colorimetric cytotoxicity assay
The cytotoxic effect of E. hirta extract on several cell lines was evaluated by micro-culture tetrazolium assay (MTT). The multiple concentrations of E. hirta extract were used and IC50 doses were calculated from the dose–response curve. The results of the cytotoxicity evaluation against cervical, colon and breast cancer cell lines, and Vero cell of E. hirta extract are shown in . Euphorbia hirta extract exhibited no cytotoxic effect on Vero cell line, with an IC50 value of 122.85 µg/mL. On the contrary, E. hirta extract showed selective cytotoxicity effects on various cancerous cell lines including cervical, colon, and breast cancer cell lines used in this study. It was also shown that all treatments displayed a percentage of growth inhibition activity in a dose-dependent manner. The cytotoxic effect of E. hirta extract was expressed as the half maximal inhibitory of concentration (IC50). Since the lower the IC50 value the higher the cytotoxic effect of the sample, IC50 growth inhibition after incubation 24 h of E. hirta extract against Vero, Hela, MCF-7, and HT29 cells was determined for which the values were 122.85 ± 4.60, >200, 25.26 ± 0.18, and 136.00 ± 1.56 μg/mL, respectively (). This study showed that MCF-7 cells had the lowest IC50 value. The Vero cells were used for comparisons between normal and cancer-derived cell models in this study and the result showed that the extract does not show any apparent cytotoxicity against the normal cells. Euphorbia hirta inhibited the proliferation of MCF-7 cells in a dose- and time-dependent manner () with the significant cytotoxic effect at 24 h with a concentration of 25.26 μg/mL. Doxorubicin hydrochloride, a standard therapeutic drug, was used as a positive control in this experiment and showed great toxicity towards the entire cancer cell lines tested. All cell lines tested were exhibited anticancer activities in time-dependent manner except HT29 does not exhibited time-dependent manner anticancer activity (). In MTT assay, formazan accumulation directly reflects the mitochondrial activity in the live cells, which is an indirect measurement for the cell viability (Mosmann, Citation1983). Since E. hirta extract showed better cytotoxicity activity against MCF-7 cells, it was used for further antiproliferative investigation.
Figure 2. The viability of various cells after exposed to the different concentrations of E. hirta extract for 24 h as determined by using MTT assay.
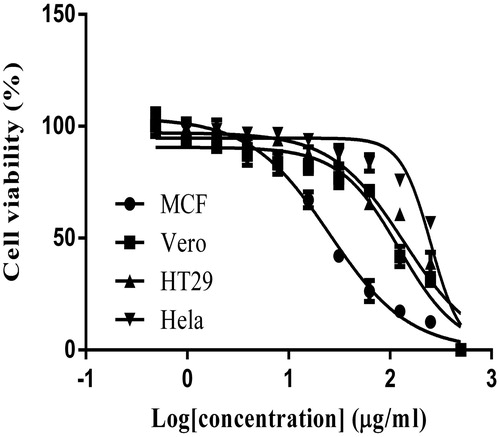
Table 1. IC50 values of E. hirta extract towards MCF-7, Hela, HT29, and Vero cells at 24, 48, and 72 h as determined by using the MTT assay.
Light microscopic studies
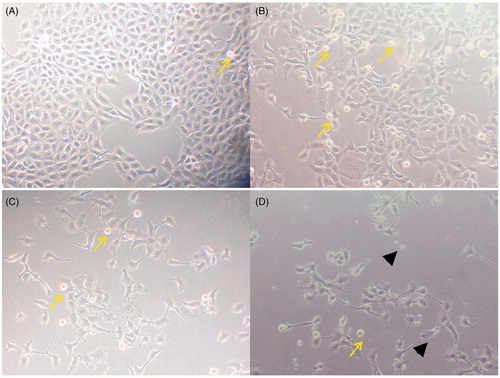
Morphological investigation of apoptosis by confocal microscopy revealed that E. hirta extract at a concentration of 25 μg/mL induced cell death in MCF-7 cell lines by apoptosis, as shown in . Phenotypically, apoptosis is characterized by cell shrinkage, chromatin condensation, plasma membrane blebbing, DNA fragmentation, and collapse of the cell into small fragments (apoptotic bodies). The phase contrast images showed that E. hirta extract decreased the number of cells and induced cell shrinkage and cell detachment from the monolayer surface compared with untreated cells. Apoptotic bodies, which appeared to be round or oval masses of cytoplasm, could be seen. shows an increase in the number of apoptotic cells corresponding to the length of incubation with E. hirta extract.
Figure 3. Morphological changes of MCF-7 cells treated with 25 μg/mL of E. hirta extract for (B) 24 h, (C) 48 h, and (D) 72 h viewed under an inverted light microscope. Control (A) was also included (200 × magnification). Arrows showed cell shrinkage and nuclear condensation due to apoptosis. Arrowheads showed the presence of apoptotic bodies.
Morphology of apoptosis by AO/PI staining
AO/PI staining was performed to determine if exposure to E. hirta extract causes cell death by apoptosis in MCF-7. In the AO/PI staining, the live cells with intact membranes have a uniform green color in their nuclei. Early apoptotic cells have chromatin condensation with bright green colored nuclei. Late apoptotic cells have bright orange areas of condensed chromatin in the nucleus that distinguish them from necrotic cells, which have a uniform orange color (Cury-Boaventura et al., Citation2004). shows the MCF-7 cells stained with acridine orange and propidium iodide together. It was found that the untreated control cells were mostly green with an intact nucleus (). As shown in , the extract was potent in inducing apoptosis in MCF-7 cells in a time-dependent manner. AO/PI analysis indicated that E. hirta extract was cytotoxic towards MCF-7 cells through apoptosis when treated with IC50 concentration. In the AO/PI analysis, cells treated with IC50 value of E. hirta extract displayed orange colored nuclei, which further confirmed the induction of late apoptosis in MCF-7 cells by E. hirta extract. This finding was characterized by membrane blebbing, nuclear shrinkage, and significant DNA fragmentation. Necrotic cells were stained red by PI, which penetrated the nuclear matter where the integrity of the cell membrane was compromised.
Figure 4. (a) AO/PI staining of MCF-7 cells treated with 25 μg/mL of E. hirta extract for 24 (B), 48 (C), and 72 (D) h, viewed under a fluorescence microscope. Control (A) was also included (200 × magnification). Untreated cells showing uniformly stained nuclei representing viable cells (A). Appearance of membrane blebs, cell shrinkage, chromatin condensation, and DNA fragmentation are signs of apoptosis of cells (B, C, and D). V, viable cell; AP, apoptotic cell; N, necrotic cell; (b) assessment of frequencies of viable, apoptotic, and necrotic cells in E. hirta extract (25 μg/mL) treated MCF-7 cancer cells at various incubation times. Values are mean ± SD (n = 3). Asterisk (*) indicates a significant p value < 0.05, as compared with controls.
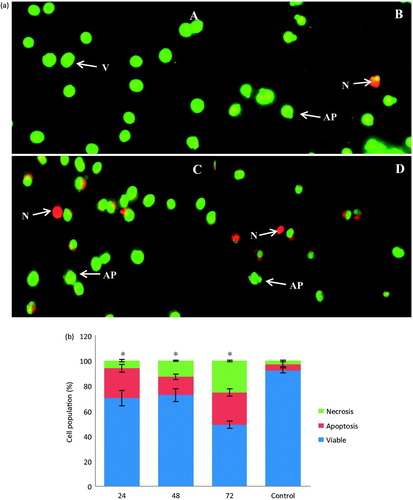
shows the proportion of apoptotic, necrotic, and viable cells in a population of MCF-7 cells treated with IC50 concentration of E. hirta extract for different time intervals. The apoptotic and necrotic cell populations increased with the time of incubation with E. hirta extract when compared with the control cells. After 24 h of treatment, the apoptotic cell population increased from 4.74% to 23.66% compared with the untreated cell population (p < 0.05). However, after 48 h of treatment, the apoptotic cell population was reduced to 14.54% while the necrotic cells increased to 12.78%. This situation may be due to the progression of the apoptotic cells into secondary necrotic cells after a certain period. The term secondary necrosis refers to a process in which late stage apoptotic cells that failed to be engulfed by phagocytes or neighboring cells undergo necrosis. Secondary necrosis, thus, is a post-apoptotic event. This process is seen in cultured cells that are undergoing cell death by apoptosis in vitro, induced, for example, by the absence of survival factor signals or activation of death receptors by different lethal signals. These cells, in the absence of phagocytic cells that could engulf them, ultimately cease to be metabolically active, lose membrane integrity, and release their cytoplasmic contents into the culture medium (Aderem & Underhill, Citation1999). After 72 h of treatment, the apoptotic cell population increased to 25.74%, while the necrotic cell population increased to 25.20% (p < 0.05).
LDH leakage assay
The cell membrane damaged by the cytotoxic agent allowed the release of intracellular LDH molecules into the culture medium. Therefore, the LDH leakage indirectly reflects the compromised cell membrane integrity, which is associated with necrosis. Necrosis is another form of cell death that will provoke an inflammatory response of surrounding cells through the leakage of intracellular contents. As shown in , the percentage of LDH leakage in E. hirta induced MCF-7 cells remained at low levels (<25%), when compared with the level of the control cells (15%). This observation is explained since many of E. hirta extract-treated cells had morphological features of apoptosis rather than necrosis. Moreover, the rapid loss of plasma membrane integrity could not be used as a marker to distinguish apoptotic from the necrotic mode of cell death (Pozarowski et al., Citation2003). It should be noted, however, that the integrity of the plasma membrane was also compromised at a late stage of apoptosis, the stage that some authors define as “secondary necrosis” (Pozarowski et al., Citation2003). This suggests that E. hirta extract induces minimal necrotic effects and has an negligible influence on the LDH leakage.
Figure 5. The levels of LDH released into the medium of untreated control and E. hirta extract-treated (50, 25, 12.5, 6.25, 3.125, and 1.5625 μg/mL) MCF-7 cells for 24 h. Values are mean ± SD (n = 3).
ROS assay
The ROS generation is commonly postulated to play a key role in apoptosis. Therefore, we investigated E. hirta extract-dependent intracellular ROS generation in MCF-7 cells. The DCF fluorescence intensity reached a peak in the cells treated with only H2O2. However, treatment with E. hirta extract did not increase the levels of the fluorescence intensity in the MCF-7 cells compared with the control (). There was no significant difference in the intracellular ROS generation between the cells exposed to E. hirta extract and the negative control cells. This suggests that E. hirta extract has an negligible influence in generating intracellular ROS. This finding indicates that E. hirta extract might exert its cytotoxicity effect via a ROS-independent mechanism in MCF-7 cells.
Figure 6. Effect of E. hirta extract on intracellular ROS production by MCF-7 cells.
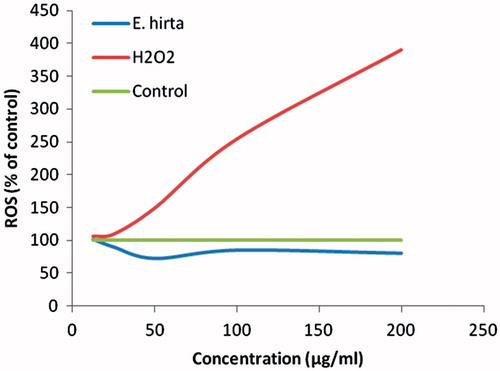
ROS plays a very important role in apoptosis induction under both physiological and pathological conditions (Li et al., Citation2006). In order to further demonstrate if ROS production is involved in apoptosis induced by E. hirta extract, the intracellular peroxide level was determined by measuring the fluorescence intensity of DCF. ROS are continually generated and eliminated in the biological system, and play significant roles in a variety of normal biochemical functions and abnormal pathological processes (Fridovich, Citation1978). Several anticancer agents, including anthracyclines, cisplatin, bleomycin, and irradiation, which are currently used for cancer treatment, have been shown to cause increased intracellular ROS generation (Chan & Yu, Citation2000; Hug et al., Citation1997; Miyajima et al., Citation1997; Serrano et al., Citation1999). However, some studies have reported that anticancer agents, such as quercetin and emodin, induce apoptosis in HL-60 cells through a ROS-independent mechanism (Chen et al., Citation2002). In addition, the increase of ROS generation in cancer cells can further stimulate cell proliferation, cause DNA mutations, and promote genetic instability and the emergence of drug resistant cells (Pelicano et al., Citation2004). The results in the present study indicate that E. hirta extract did not increase the levels of intracellular ROS even at high concentrations. This data demonstrate that E. hirta extract-induced apoptosis is independent of ROS production. Phytochemicals, such as flavonoids and triterpenoids, can act as antioxidants in cells by modulating the activity of enzymatic and non-enzymatic cellular antioxidants as well as by activating the phase I (activating) enzymes and phase II (detoxifying) enzymes involved in xenobiotic metabolism (Ramos et al., Citation2010). The free radical scavenging and antioxidant property of E. hirta extract have been recently reported by Basma et al. (Citation2011a). Moreover, natural products have been constantly described as possessing prooxidant and antioxidant activities. In the present study, we found that E. hirta extract itself did not alter the intracellular ROS level although apoptosis was induced. Therefore, it is interesting to investigate if ROS is involved in the differential apoptosis inducing activities of E. hirta extract.
E. hirta extract induced apoptosis in MCF-7 cells
Cell death induced by E. hirta extract was investigated for apoptotic activity by monitoring phosphatidylserine (PS) translocation using the annexin V-FITC/PI assay. Early apoptosis is characterized by the translocation of PS from the inner layer of the plasma membrane to the outer surface (Cury-Boaventura et al., Citation2004). Apoptotic cells are reflected by quantification of Annexin V-FITC binding to externalized PS. In flow cytometry analysis, Annexin V/propidium iodide (AnnV/PI) staining is based on the ability of the protein Annexin V to bind to phosphatidylserine (PS), which is externalized in the outer cell membrane leaflet upon induction of apoptosis. In viable cells, PS is located in the inner membrane leaflet, but upon induction of apoptosis it is translocated to the outer membrane leaflet and becomes available for Annexin V binding. The addition of PI enabled viable (AnnV−/PI−), early apoptotic (AnnV+/PI−), late apoptotic (AnnV+/PI+), and necrotic (AnnV−/PI+) cells to be distinguished (Baskic et al., Citation2006). The flow cytometry analysis of MCF-7 cells showed that through the treatment with E. hirta extract the cell population tended to shift from viable to apoptotic.
After treatment with 25.00 µg/mL E. hirta extract for 24, 48, and 72 h, the early apoptotic rates were 34.97, 32.4, and 38.03%, respectively, compared with the control 13.7% (). Furthermore, non-significant changes in the percentage of late stage apoptotic cells for E. hirta extract-treated group were observed at 24, 48, and 72 h of incubation. These results demonstrate the ability of E. hirta extract to exert apoptosis, particularly in early stage apoptosis in MCF-7 cells.
Figure 7. Annexin V/PI double-staining assay of MCF-7 cells treated with E. hirta extract (25 μg/mL) at (A) 0 h, (B) 24 h, (C) 48 h, and (D) 72 h. The Y-axis represents the PI-labeled population, whereas the X-axis represents the FITC-labeled Annexin V positive cells. The lower left portion of the fluorocytogram (An−, PI−) shows normal cells, whereas the lower right portion of the fluorocytogram (An+, PI−) shows early apoptotic cells. The upper right portion of the fluorocytogram (An+, PI+) shows late poptotic cells. The apoptotic rates of MCF-7 cells induced by E. hirta extract. *p < 0.05 and **p < 0.01 versus control of each Q, ANOVA with the Tukey post test.
Induction of DNA fragmentation by E. hirta
To confirm the induction of apoptosis, MCF-7 cells were treated with various concentrations of E. hirta extract, and DNA was isolated and analyzed by agarose gel electrophoresis. In the experiments with DNA from MCF-7 cells treated with various concentrations of E. hirta extract for 24 h, a typical ladder pattern of internucleosomal fragmentation was observed (). A significant ladder pattern of DNA fragmentation was found when MCF-7 cells were treated with 25.00 µg/mL of E. hirta extract. These results indicate that E. hirta extract caused DNA fragmentation in MCF-7 cells and that the susceptibility of MCF-7 cells to E. hirta extract-induced cell death was higher at the IC50 concentration. During apoptosis, DNA strand breaks are present and the nicks in DNA molecules can be detected via DNA fragmentation assay. It is well known that apoptosis is involved in the activation of endonucleases, which results in the formation of DNA fragmentation; this can be seen through electrophoretic examination (Eastman & Barry, Citation1992). Internucleosomal DNA fragmentation, as determined by agarose gel electrophoretic analysis, in this study, also supported the progression of apoptosis in E. hirta extract treated MCF-7 cells. Normally, DNA fragmentation during necrosis appears to be less extensive, as reflected by only a minor decrease in the cellular DNA content.
Figure 8. DNA fragmentation of MCF-7 cells exposed to E. hirta. Fragmentations of genomic DNA in MCF-7 cells were treated for 24 h with 50, 25, and 12.5 µg/mL of E. hirta. DNA laddering formation was viewed on ethidium bromide-stained gel (2%) and photographed by UV illumination. Marker, marker 100 bp; Control, DMSO control; DOX, doxorubicin hydrochloride.
Cell cycle
After MCF-7 cells were incubated with 12.50, 25.00, and 50.00 µg/mL of E. hirta extract, they were isolated to examine the effects on cell cycle and apoptosis by flow cytometry. The results indicate that E. hirta extract induced S phase arrest (). After 24 h treatment with various concentrations of E. hirta extract, a significant increase in the number of cells arrested at the S growth phase was observed. Increasing the concentration of E. hirta extract from 12.50 to 50.00 μg/mL caused a significant increase in the S phase. Although the increase in the percentage of cells in S is not large, the results are statistically significant (p < 0.05) and are consistent. As shown in , E. hirta extract caused a comparable decrease in the G0/G1 phase in a dose-dependent manner. In short, E. hirta extract caused S cell-cycle arrest in a concentration-dependent manner in MCF-7 cells, which was accompanied by a proportional decrease in the percentage of cells in the G0/G1 phase. Furthermore, the treatment of E. hirta extract caused a decreased in the sub-G1 cells population. The percentage of cells in G2/M phase was increased with concentration till 25.00 µg/mL and the results were statistically significant (p < 0.05) compared with control. The results indicated that E. hirta arrested the cell cycle of the MCF-7 cells in the S and G2/M phases and induced apoptosis after treatment for 24 h. However, the cell-cycle arrest analysis showed that treatment with the positive control doxorubicin caused cell arrest at the G0/G1 phase.
Figure 9. Euphorbia hirta extract induces cell cycle arrest in MCF-7 cells. Cells were treated with DMSO (negative control), doxorubicin at 0.939 μg/mL (positive control) and E. hirta extract (25.000 μg/mL) for 24 h. Flow cytometric analysis was performed for cell-cycle distribution. The DNA content was evaluated with propidium iodide (PI) staining and fluorescence measured and analysed. (a) Representative flow cytometry graph for each treated or untreated groups. (b) Percentage of cells in various phases in histograms. Data represent mean ± SD of triplicate, *p < 0.05 as compared with the control group.
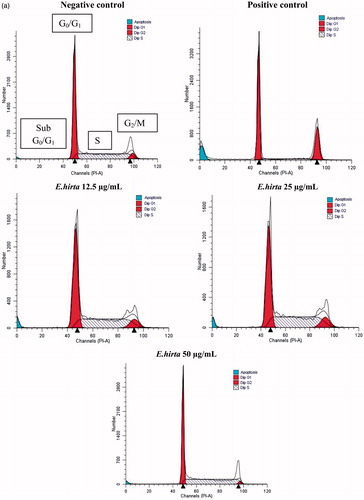
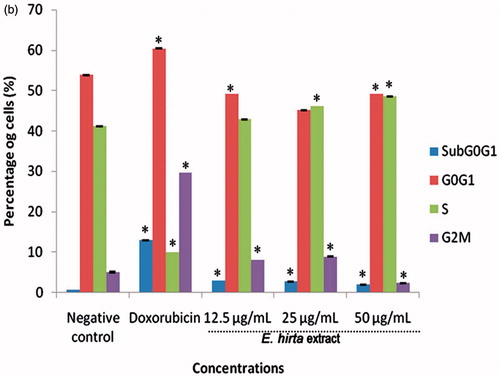
Many anticancer agents arrest the cell cycle at the G0/G1, S, and G2/M phase and then induce apoptosis (Martínez et al., Citation1987; Murray, Citation2004; Torres & Horwitz, Citation1998). Hence, the effect of E. hirta on the cell cycle was determined to identify the mechanism of action of E. hirta inhibiting MCF-7 cell growth. Flow cytometry analysis of this study showed that E. hirta extract can cause S phase arrest in a dose-dependent manner. Cell cycle and apoptosis play important roles in the regulatory mechanisms of cell growth. Cell-cycle checkpoints ensure the maintenance of genomic integrity by protecting dividing cells from the potentially fatal consequences of DNA damage. The detection of unrepaired DNA damage may force the cells to withdraw from the cell cycle or die by apoptosis so that cells do not replicate chromosomes with irreparable damage (Nakanishi et al., Citation2006). For the first time, this study shows that cell-cycle disruption is the basis of anti-proliferative action of E. hirta extract. The inhibitory concentration of the E. hirta extract acted by specifically blocking the cell cycle at S-phase and prevented the cell from entering the proliferative phase. Moreover, S phase transition provides an efficient check point in cell-cycle progression that can severely affect the proliferation of the MCF-7 cell population. Furthermore, aberrant constitutive E2F-1 activity during S phase arrest has been shown to result in apoptosis (Krek et al., Citation1995). Though an elegant study previously demonstrated that increased levels of E2F-1 activity could contribute to the selective killing of transformed cells when treated with flavopiridol during the S phase (Jiang et al., Citation2003). This suggests an association between the sensitivity of cells in the S phase and the apoptotic response through E2F activity. Moreover, retinoblastoma gene product (Rb) governs entry into S phase. Hypophosphorylated Rb forms a complex with transcription factors, E2F. When Rb is inactivated by cyclin-dependentkinases (CDK) 2 -, CDK4-, or CDK6-mediated phosphorylation, E2F transcription factors are released, resulting in progression into S phase (Shah & Schwartz, Citation2001). The cell-cycle analysis also showed a decrease in the sub-G1 cells population with a very low percentage of cells. A cell must have lost enough DNA in order to appear in the sub-G1 area (Darzynkiewicz et al., Citation1997). Therefore, if a cell enters apoptosis from the S or G2/M phase of the cell cycle, they may not appear in the sub-G1 area. Thus, apoptotic cells from E. hirta extract treatment showed a decreased percentage of sub-G1 cells population. In addition, during cell cycle, the G2/M checkpoint is a possible target for anticancer therapy. It prevents DNA-damaged cells from entering mitosis and allows for the repair of DNA damage. Moreover, defects in the G2/M arrest checkpoint may allow a damaged cell to enter mitosis and undergo apoptosis, and efforts to enhance this effect may increase the cytotoxicity of chemotherapy (Wang et al., Citation2009). For this reason, our results suggest that E. hirta extract induced cell-cycle S and G2/M phases arrest and apoptosis in MCF-7.
Caspase-2, 3, 6, 8, and 9 activities
shows the results of colorimetric analysis of caspase-2, 3, 6, 8, and 9 in MCF-7 cells treated with E. hirta extract. It reveals that E. hirta extract significantly increased the caspase-2 and 6 activities compared with the untreated control, suggesting a possible relation between E. hirta extract-induced apoptosis and the activation of caspase-2 and 6. However, there was no significant effect on caspase-3 activity by E. hirta extract although an increase in caspase-8 and 9 activities was observed compared with the uninduced control.
Figure 10. Caspases activity in MCF-7 cells was analyzed using caspase colorimetric protease assay kit. Increases in caspases activity in cells after treatment with 25.00 µg/mL of E. hirta extract for 24 h and was determined by comparing the results with control. The values are expressed as means ± SEM of at least two independent experiments. Significant differences compared between control and treated cells (*p < 0.05).
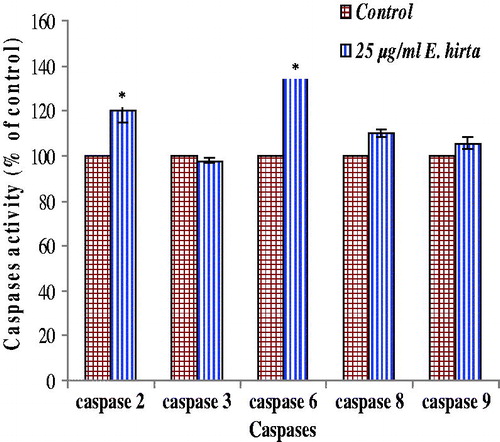
Apart from the cell-cycle regulation, increasing evidence suggests that caspase family proteases play an important role in the mechanism of apoptosis. In apoptosis, caspase are indispensable enzymes that control the pathway. Caspase activation analysis of this study showed activation of caspase-2, 6, 8, and 9 by E. hirta extract but no activation of caspase 3 was observed. It has been shown that the MCF-7 cell line was established from the breast cancer of a Caucasian woman, lack caspase-3 activity due to a point mutation in the gene coding for this protein (Liu et al., Citation1996; Janicke et al., Citation1998; Srinivasula et al., Citation1996). The caspase 3 subfamily consists of caspases 3, 6, 7, 8, 9, and 10. Of these, those with large prodomains (i.e., caspases 8, 9, and 10) are considered initiator caspases and those with a small prodomain (i.e., caspases 3, 6, and 7) are considered effector or executioner caspases (Srinivasula et al., Citation1996). Therefore, caspase-6, 8 and 9, which is a member of the caspase-3 subfamily, may be involved in apoptosis in caspase-3-deficient MCF-7 cells, which deserve further investigation. Caspase-2 is unclear whether it should be considered an initiator or an effector caspase. Although caspase-2 has been proposed to act upstream of mitochondria and to be activated within the p53-induced protein with a death domain (PIDD)–RIP-associated Ich-1/Ced-3-homologue protein with a death domain (RAIDD)-caspase-2 complex (PIDDosome), it may also be activated downstream of caspase-3 (Inoue et al., Citation2009). On the contrary, caspase-6 is generally considered to be an effector caspase. Caspase-6 cleaves several substrates including nuclear lamins, focal adhesion kinase, nuclear mitotic apparatus protein (NUMA), and the nuclear matrix protein, special AT-rich binding protein-1 (SATB1). Most caspase-6 substrates, except for lamin A/C and SATB1, can be cleaved by other caspases. In the absence of caspase-6, cells with lamin A do not undergo complete nuclear condensation during the execution phase of apoptosis (Inoue et al., Citation2009). In addition, it has been verified that caspase-3-deficient human breast cancer cells, such as MCF-7 cells, are resistant to apoptotic stimuli and are imperfect in response to chemotherapy and radiotherapy (Chang et al., Citation2011). Unfortunately, about 75% of breast cancer tissues are down-regulated by or lack caspase-3 expression, which is considered to be associated with the resistance to chemotherapy and radiotherapy in breast cancer patients (Chang et al., Citation2011). Therefore, developing new therapeutic agents from E. hirta extract, which exhibit the mechanism of action through the caspase-3 independent pathway, is strongly recommended.
Fractionation and identification of cytotoxic fraction from E. hirta
To identify the active fraction in E. hirta extract which possesses cytotoxic activity against MCF-7 cell line, the extract was partitioned with various solvent systems namely hexane, ethyl acetate, and butanol and chromatographically fractionated. Among these fractions, two HF subfractions (namely HFsub4 and HFsub7), four EAF subfractions (namely EAFsub1, EAFsub6, EAFsub7, and EAFsub8) and two BF subfractions (namely BFsub8 and BFsub9) showed cytotoxic activity on MCF-7 cells. Among these subfractions, HFsub4 showed highest activity on MCF-7 cells with an IC50 value of 10.01 µg/mL (). The IC50 values indicated that the anticancer activity of the HFsub4 was higher than HF and E. hirta extract against MCF7 cell lines. Thus the active fraction is considered to be contained the candidate compound for anticancer drug.
Table 2. IC50 values of E. hirta extract various isolated fractions towards MCF-7 at 24 h as determined by using the MTT assay.
Conclusions
In summary, the present results indicate that E. hirta extract induced apoptosis through the caspase-3 independent pathway and S and G2/M phases arrest in MCF-7 cells, which was further confirmed by characteristic morphological changes and DNA fragmentation appearance. However, more studies will be needed to examine the involvement of multiple cyclin-dependent kinases in the phosphorylation of E2F-1 and the state of retinoblastoma (Rb) phosphorylation to determine the regulation of E. hirta-induced apoptosis during S phase arrest in breast cancer cells. Bioassay has led to identification of HFsub4 fraction as the cytotoxic fraction in E. hirta extract. Therefore, our results suggest that E. hirta extract with HFsub4 fraction could be a good candidate for developing anticancer agents.
Declaration of interest
The authors report that they have no conflicts of interests. Kwan Yuet Ping is supported by the MyPhD fellowship from the Ministry of Higher Education of the Malaysian Government. This work was partly supported by UM-MoHE HIR Grant (No. UM-MoHE HIR H-18001-C0020) from the University of Malaya, Malaysia.
References
- Aderem A. (1999). Underhill DM. Mechanisms of phagocytosis in macrophages. Ann Rev Immunol 17:593–623
- Baskic D, Popovic S, Ristic P, et al. (2006). Analysis of cycloheximideinduced apoptosis in human leukocytes: Fluorescence microscopy using annexin V/propidium iodide versus acridin orange/ethidium bromide. Cell Biol Int 30:924–32
- Basma AA, Zakaria Z, Latha LY, et al. (2011a). Antioxidant activity and phytochemical screening of the methanol extracts of Euphorbia hirta L. Asian Pac J Trop Med 5:386–90
- Basma AA, Zuraini Z, Sasidharan S. (2011b). A transmission electron microscopy study of the diversity of Candida albicans cells induced by Euphorbia hirta L. leaf extract in vitro. Asian Pac J Trop Biomed 1:20–2
- Chan WH, Yu JS. (2000). Inhibition of UV irradiation-induced oxidative stress and apoptotic biochemical changes in human epidermal carcinoma A431 cells by genistein. J Cell Biochem 78:73–84
- Chang HC, Chen ST, Chien SY, et al. (2011). Capsaicin may induce breast cancer cell death through apoptosis-inducing factor involving mitochondrial dysfunction. Hum Exp Toxicol 30:1657–65
- Chen YC, Shen SC, Lee WR, et al. (2002). Emodin induces apoptosis in human promyeloleukemic HL-60 cells accompanied by activation of caspase 3 cascade but independent of reactive oxygen species production. Biochem Pharmacol 64:1713–24
- Cury-Boaventura F, Pomp C, Curi R. (2004). Comparative toxicity of oleic acid and linoleic acid on Jurkat cells. Clin Nutr 23:721–32
- Darzynkiewicz Z, Juan G, Li X, et al. (1997). Cytometry in cell necrobiology: Analysis of apoptosis and accidental cell death (necrosis). Cytometry 27:1–20
- Dobashi Y, Takehana T, Ooi A. (2003). Perspectives on cancer therapy: Cell cycle blockers and perturbators. Curr Med Chem 10:2549–58
- Eastman A, Barry MA. (1992). The origins of DNA breaks: A consequence of DNA damage, DNA repair or apoptosis? Cancer Invest 10:229–40
- Fridovich I. (1978). The biology of oxygen radicals. Science 201:875–80
- Gerl R, Vaux DL. (2005). Apoptosis in the development and treatment of cancer. Carcinogenesis 26:263–70
- Hug H, Strand S, Grambihler A, et al. (1997). Reactive oxygen intermediates are involved in the induction of CD95 ligand mRNA expression by cytostatic drugs in hepatoma cells. J Biol Chem 272:28191–3
- Inoue S, Browne G, Melino G, et al. (2009). Ordering of caspases in cells undergoing apoptosis by the intrinsic pathway. Cell Death Differ 16:1053–61
- Janicke RU, Sprengart ML, Wati MR, et al. (1998). Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem 273:9357–60
- Jiang J, Matranga CB, Cai D, et al. (2003). Flavopiridol-induced apoptosis during S phase requires E2F-1 and inhibition of cyclin A-dependent kinase activity. Cancer Res 63:7410–22
- Johnson PB, Abdurahman EM, Tiam EA, et al. (1999). Euphorbia hirta leaf extracts increase urine output and electrolytes in rats. J Ethnopharmacol 65:63–9
- Hassan M, Watari H, AbuAlmaaty A, et al. (2014). Apoptosis and molecular targeting therapy in cancer. Biomed Res Int 2014:150845
- Kim H, Choi JY, Lee JM, et al. (2008). Dexamethasone increases angiopoietin-1 and quiescent hematopoietic stem cells: A novel mechanism of dexamethasone-induced hematoprotection. FEBS Lett 582:3509–14
- Kraft K. (2009). Complementary/alternative medicine in the context of prevention of disease and maintenance of health. Prev Med 49:88–92
- Kumar S, Malhotra R, Kumar D. (2010). Euphorbia hirta: Its chemistry, traditional and medicinal uses, and pharmacological activities. Pharmacogn Rev 4:58–61
- Krek W, Xu G, Livingston DM. (1995). Cyclin A-kinase regulation of E2F-1 DNA binding function underlies suppression of an S phase checkpoint. Cell 83:1149–58
- Lanhers MC, Fleurentin J, Dorfman P, et al. (1991). Analgesic, antipyretic and anti-inflammatory properties of Euphorbia hirta. Planta Med 57:225–31
- Li JJ, Tang Q, Li Y, et al. (2006). Role of oxidative stress in the apoptosis of hepatocellular carcinoma induced by combination of arsenic trioxide and ascorbic acid. Acta Pharmacol Sin 27:1078–84
- Liu X, Kim CN, Yang J, et al. (1996). Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome C. Cell 86:147–57
- Mamun-Or-Rashid ANM, Shohel M, Nayeem MT, et al. (2013). Compendium ethnopharmaceutical review on Euphorbia hirta. Ayurpharm Int J Ayur Alli Sci 2:14–21
- Martínez V, Barberá O, Sánchez-Parareda J, et al. (1987). Phenolic and acetylenic metabolites from Artemisia assoana. Phytochemistry 26:2619–26
- Mishell BB, Shiiqi SM, Henry C. (1980). Selected methods. In: Mishell BB, Shiiqi SM, eds. Cellular Immunology. San Francisco: Freeman, 21–2
- Miyajima A, Nakashima J, Yoshioka K, et al. (1997). Role of reactive oxygen species in cis-dichlorodiammineplatinum-induced cytotoxicity on bladder cancer cells. Br J Cancer 76:206–10
- Mok DKW, Chau FT. (2006). Chemical information of Chinese medicines: A challenge to chemist. Chemom Intell Lab Syst 82:210–17
- Mosmann T. (1983). Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63
- Murray AW. (2004). Recycling the cell cycle: Cyclins revisited. Cell 116:221–34
- Nakanishi M, Shimada M, Niida H. (2006). Genetic instability in cancer cells by impaired cell cycle checkpoints. Cancer Sci 97:984–9
- Pelicano H, Carney D, Huang P. (2004). ROS stress in cancer cells and therapeutic implications. Drug Resist Updates 7:97–110
- Pozarowski P, Halicka DH, Darzynkiewicz Z. (2003). NF-κB Inhibitor sesquiterpene parthenolide induces concurrently atypical apoptosis and cell necrosis: Difficulties in identification of dead cells in such cultures. Cytometry Part A 54:118–24
- Rajeh BM, Zuraini Z, Sasidharan S, et al. (2010). Assessment of Euphorbia hirta L. leaf, flower, stem and root extracts for their antibacterial and antifungal activity and brine shrimp lethality. Molecules 15:6008–18
- Ramos AA, Pereira-Wilson C, Collins AR. (2010). Protective effects of ursolic acid and luteolin against oxidative DNA damage include enhancement of DNA repair in Caco-2 cells. Mutat Res 692:6–11
- Serrano J, Palmeira CM, Kuehl DW, et al. (1999). Cardioselective and cumulative oxidation of mitochondrial DNA following subchronic doxorubicin administration. Biochim Biophys Acta 1411:201–5
- Shah MA, Schwartz GK. (2001). Cell cycle-mediated drug resistance: An emerging concept in cancer therapy. Clin Cancer Res 7:2168–81
- Shih MF, Cherng JY. (2012). Potential applications of Euphorbia hirta in pharmacology. In: Omboon V, Suleiman MO, eds. Drug Discovery Research in Pharmacognosy. Croatia: InTech, 165–80
- Srinivasula SM, Ahmad M, Fernandes-Alnemri T, et al. (1996). Molecular ordering of the Fas-apoptotic pathway: The Fas/APO-1 protease Mch5 is a CrmA-inhibitable protease that activates multiple Ced-3/ICE-like cysteine proteases. Proc Natl Acad Sci 93:14486–91
- Torres K, Horwitz SB. (1998). Mechanisms of taxol-induced cell death are concentration dependent. Cancer Res 58:3620–6
- Tyagi AK, Singh RP, Agarwal C, et al. (2002). Silibinin strongly synergizes human prostate cancer DU145 cells to doxorubicin-induced growth inhibition, G2-M arrest, and apoptosis. Clin Cancer Res 8:3512–19
- Wang Y, Ji P, Liu J, et al. (2009). Centrosome-associated regulators of the G2/M checkpoint as targets for cancer therapy. Mol Cancer 8:8
- Yuet Ping K, Darah I, Chen Y, et al. (2013). Acute and subchronic toxicity study of Euphorbia hirta L. methanol extract in rats. Biomed Res Int 2013:182064