
Abstract
Context Hydrogen peroxide (H2O2)-induced damage in the lens epithelium leads to cell death and cataract. Semiaquilegia adoxoides (DC.) Makino (Ranunculaceae), a folk medicine of Hmong (an ethnic group of China), has been traditionally used to treat cataract; however, the underlying molecular mechanism is yet to be uncovered.
Objective This study aimed to investigate whether the n-butanol extract of S. adoxoides (nSA) is effective against the H2O2-induced oxidative stress in human lens epithelial (HLE) cells.
Materials and methods Human lens epithelial (SRA 01/04) cells were stimulated by H2O2 (250 μM) in the presence or absence of nSA. The antioxidant effects of nSA were determined in terms of cell viability (MTT assay), apoptosis (AnnexinV/PI staining), radical scavenging capability (various enzymatic assays), loss of mitochondrial membrane potential (Rhodamine 123 staining), expression of apoptotic markers including caspase-3 and caspase-9 and the change of Bcl-2/Bax ratio (western blot) in the HLE cells.
Results The results showed that pretreatment of nSA (250, 500 and 1000 μg/mL) markedly reduced H2O2-induced cellular apoptosis and malondialdehyde accumulation, but elevated the activities of total superoxide dismutase, catalase, glutathione peroxidase. Thus, the total antioxidative capability was enhanced upon the nSA treatment meanwhile the loss of mitochondrial membrane potential was prevented. Moreover, nSA at concentrations of 250, 500 and 1000 μg/mL also significantly suppressed the activation of caspase-3 and -9, and increased the Bcl-2/Bax ratio in the HLE cells.
Discussion and conclusion Our findings suggested that nSA is a potential prophylactic agent in the prevention of cataractogeneis.
Introduction
Cataract is the leading cause of blindness among elderly populations and causes enormous social and economic burdens to many countries (Salm et al. Citation2006; Tang et al. Citation2011). Although cataract surgery, an outpatient procedure, is considered an effective therapy to cataract patients, risks and complications are often associated, especially in aged people (Meekins & Afshari Citation2012). According to a previous report, if the onset of cataract can be impeded for 10 years in the patients, half of the cataract surgeries are estimated as unnecessary as the average age for cataract surgery is approximately 75 years old (Spector & Garner Citation1981). Our transparent ocular lens is indeed composed of fibre cells, which are derived from epithelia. Once they are damaged, they do not self-renew, and become permanently impaired (Zheng et al. Citation2010). The single layer of epithelial lining on the anterior surface of lens is critical for the maintenance of metabolic homeostasis and transparency of the entire lens (Paron et al. Citation2004; Ou et al. Citation2012). Damage caused to these epithelial cells result in the degradation of corneal proteins and ultimately elicit cataract formation. Accumulating evidence showed that oxidative stress is a major contributor to the initiation and progression of cataract (Truscott Citation2005; Li et al. Citation2009; Yao et al. Citation2009; Tang et al. Citation2011). In fact, we have a variety of self-defence mechanisms, for instance, enzyme-dependent systems such as superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GSH-Px) (Giblin Citation2000; Zheng et al. Citation2010) of our corneal lens for defending against internal and/or external stressors such as oxidative stress and ultra-violet rays. However, the overwhelmed generation of oxidative stress triggers epithelial apoptosis, hence formation of cataract is initiated (Mok et al. Citation2014). Protection against oxidative stress in human lens epithelial cells is therefore an important strategy to prevent or postpone the formation of cataract.
Semiaquilegia adoxoides (DC.) Makino (Ranunculaceae), also known as Tian-Kui-Zi, is a perennial plant (Su et al. Citation2004) mainly distributed in provinces, such as Guizhou, Sichuan, Hubei and Hunan, along the middle and lower regions of the Yangtze River in China. Semiaquilegia adoxoides has been widely used for the treatment of snakebite, bruises, tonsillitis, mastitis, scrofula and even cancers (Niu et al. Citation2006). In the Hmong population of China, the juice from plants of S. adoxoides has long been used to treat cataract (Guiyang Citation1959). In our previous study, we administered the ethanol extract of S. adoxoides to rats with d-galactose-induced cataract. Our results showed that the eye drops prepared from the ethanol extract of S. adoxoides profoundly improved the opacification of the lens in those rats with d-galactose-induced cataract. To the S. adoxoides-treated rats, we also demonstrated that the activities of total-SOD (T-SOD) in their sera were increased while the contents of corneal malondialdehyde (MDA) were decreased (Hou et al. Citation2012). However, the effect of S. adoxoides on oxidative damage in lens epithelia was not evaluated. In our preliminary work, we screened the protective effects of different fractions (petroleum ether, ethyl acetate, n-butanol and remaining aqueous fractions) of S. adoxoides on H2O2-induced oxidative damage in human lens epithelial cells (SRA 01/04). We found that the n-butanol extract of S. adoxoides showed the most potent protective effect against H2O2-induced oxidative stress in HLE cells (data not shown). In the present study, we investigated the effects of n-butanol extract of S. adoxoides (nSA) on H2O2-induced oxidative stress and the associated molecular mechanisms in the HLE cells.
Materials and methods
The aerial part of S. adoxoides was collected from Xifeng city, Guizhou province, China, in July 2012. The herb was authenticated by Professor Qing-de Long (School of Pharmacy, Guiyang Medical University, Guiyang, Guizhou province, China). A voucher specimen was stored at Laboratory of Medicinal Plant and Pharmacognosy, School of Pharmacy, Guiyang Medical University, Guiyang, China (No. P120703). Fetal bovine serum (FBS), 0.25% trypsin and DMEM/F12 medium were obtained from Gibco (Grand Island, NY). Dimethylsulphoxide (DMSO), H2O2, rhodamine123, cocktail of protease inhibitors and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma Chemical Co. (St. Louis, MO). Anti-Bax, anti-Bcl-2, anti-actin, anti-mouse and anti-rabbit IgG horseradish peroxidase (HRP) antibodies were purchased from Santa Cruz (Santa Cruz, CA). The ECL detection kit was acquired from Amersham Pharmacia (Arlington Heights, IL). Annexin V/FITC kit was purchased from eBioscience (Bender MedSystems, GmbH, Vienna, Austria). Anti-caspase-3 and anti-caspase-9 antibodies were obtained from Cell Signaling Technology (Beverly, MA). MDA, T-SOD, CAT, GSH-Px and total antioxidative capability (T-AOC) colorimetric activity assay kits were purchased from Jiancheng Bioengineering Institute (Nanjing, Jiangsu, China).
Preparation and HPLC analysis of n-butanol extract of S. adoxoides
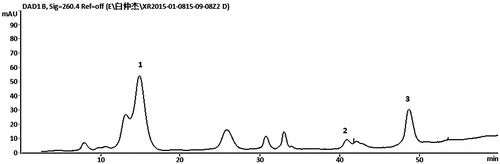
The S. adoxoides sample (100 g) was extracted twice by 10-fold 75% ethanol (v/v) for 60 min under reflux. The filtrates were combined and dried in a vacuum at 45 °C to yield the ethanol extract. Subsequently, the ethanol extract was dissolved in water and extracted by ethyl acetate to remove some non-polar compounds, and the residue was then extracted by n-butanol and concentrated under vacuum at 45 °C to yield the n-butanol extract of S. adoxoides (nSA). The extraction yield was 18.2%. The chemical profile of nSA was performed on an Agilent Series 1260 liquid chromatography (Agilent Technologies, Palo Alto, CA), equipped with a vacuum degasser, a quaternary pump, an autosampler and a DAD detector, connected to an Agilent ChemStation software (Agilent Technologies, Palo Alto, CA). The system operated at 30 °C, and a YWG C18 column (4.6 mm × 250 mm i.d., 10 μm) was used. The injection volume was 20 μL and the mobile phase flow rate was 1.0 mL/min. Solvents that constituted the mobile phase were (A) 0.05% aqueous phosphoric acid and (B) methanol. The following elution conditions were applied: 0–20 min, isocratic gradient 3% B; 20–30 min, linear gradient 3–20% B; 30–50 min, linear gradient 20–40% B; 50–60 min, linear gradient 40–50% B. Peaks were detected at 260 nm ().
Figure 1. UV-HPLC chromatographic profile of nSA. The chromatograms were obtained at 260 nm. Lithospermoside (1); chlorogenic acid (2); ferulic acid (3).
Cell culture
Human lens epithelial (SRA 01/04) cell line was obtained from The Chinese Academy of Medical Sciences (Beijing, China). The HLE cells were grown as a monolayer in DMEM/F12 medium supplemented with 15% heat-inactivated FBS, penicillin (100 U/mL) and streptomycin (100 μg/mL) at 37 °C in a humidified atmosphere of 5% CO2. The cells were routinely subcultured every 2–3 d.
Cell viability
The cells were plated at a density of 1 × 104 cells/well in 96-well microplates, and incubated with different concentrations of nSA (0–2000 μg/mL) in the presence or absence of H2O2 for 24 h (h). After the 24 h incubation, the culture medium was removed and cell viability was measured using MTT method as previously described (Xiao et al. Citation2014).
Detection of cellular apoptosis
The percentage of apoptotic cells was determined using the Annexin V/FITC apoptosis detection kit according to the manufacturer’s direction. Briefly, cells were plated and incubated in 6-well plates at a density of 1 × 106 cells/well. After exposure to H2O2 (250 μM) and/or indicated concentrations of nSA for 6 h, the cells were detached by 0.25% trypsin and washed twice with ice-cold phosphate buffered saline (PBS). Cells were collected and slightly centrifuged. Pellets were re-suspended in binding buffer (1×), and incubated with 5 μL FITC-labelled Annexin V and 10 μL propidium iodide (PI) for 15 min at room temperature in dark. The level of cell apoptosis was immediately analysed on a flow cytometer (Becton Dickinson, San Jose, CA).
Measurement of oxidative stress
HLE cells were seeded on 6-well plates at a density of 1 × 106 cells/well. After exposure to H2O2 (250 μM) and/or indicated concentrations of nSA for 24 h, cells were harvested and lysed by ultrasonic disruption on ice, and then centrifuged at 12 000 rpm for 15 min at 4 °C. The supernatant was collected for T-SOD, CAT, MDA, GSH-Px and T-AOC determination using commercial assay kits according to the manufacturer’s instruction. Protein concentrations were measured using the BCA protein assay kit. The MDA results were expressed as nmol/mg protein whereas T-SOD, CAT, GSH-Px and T-AOC were expressed as U/mg protein.
Measurement of mitochondrial membrane potential (Δψm)
The reduced cellular Δψm is considered as a marker of mitochondrial dysfunction. The level of Δψm in HLE cells was measured using Rhodamine123 (Rh123), a specific fluorescent probe for monitoring the membrane potential in living cells as previously described (Han et al. Citation2014). After exposure to H2O2 (250 μM) and/or indicated concentrations of nSA for 6 h, the cells were washed twice with PBS and incubated with Rh123 (1 μM) for 30 min at 37 °C. The fluorescent intensity was analysed with an excitation/emission wavelength at 507/529 nm on a flow cytometry (Becton Dickinson, San Jose, CA). The data were analysed by CellQuest software (BD Biosciences, San Jose, CA) and the percentage of cells with Δψm loss was calculated.
Immunoblotting analysis
Cells were plated and incubated in 6-well plates at a density of 1 × 106 cells/well. After exposure to H2O2 (250 μM) and/or indicated concentrations of nSA for 24 h, the cells were washed twice with ice-cold PBS and lysed using RIPA buffer for 30 min on ice. Protein concentrations were quantified using the BCA protein assay kit (Bradford Citation1976). Samples were diluted to a concentration of 2 mg/mL in SDS-loading buffer and boiled for 5 min. Each protein sample (50 μg) was separated by 12% SDS-PAGE and transferred onto nitrocellulose membranes. The membranes were blocked with 5% non-fat milk in 1 × Tris buffered saline buffer with 0.05% Tween-20 (TBST, pH 8.0) for 1.5 h and then incubated overnight at 4 °C with anti-Bcl-2, anti-Bax, anti-caspase-3 and anti-caspase-9 antibodies. The expression of β-actin was used to show equal protein loading. After extensive washing with TBST (3 × 10 min), the membranes were incubated with HRP-linked anti-biotin or appropriate secondary antibody in TBST with 5% non-fat milk for 1 h at room temperature, and were rewashed (TBST, 3 × 10 min). The immunobands were detected using the enhanced chemiluminescence (ECL) reaction. Quantification of protein bands was achieved by densitometric analysis using the Image-Pro Plus® software (Media Cybernetics, Inc., Rockville, MD).
Statistics analysis
The data are presented as mean value ± standard error of the mean (SEM). Statistical significances were evaluated using one-way ANOVA, followed by Duncan's multiple range tests. GraphPad Prism 5.0 software (GraphPad Software Inc., San Diego, CA) was used for all calculations, and p <0.05 was considered statistically significant.
Results
nSA inhibited H2O2-induced cell death in HLE cells
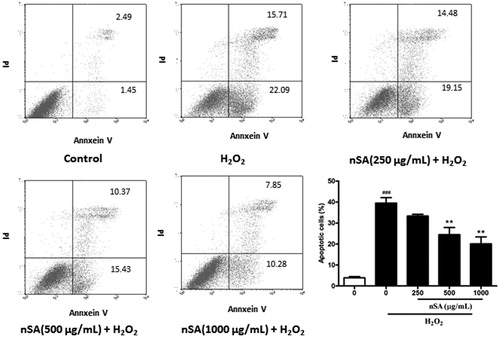
As shown in , the cytotoxicity of nSA was first evaluated using the MTT detection method. The cell viability of HLE cells was not significantly affected by treatment with any tested concentrations of nSA, indicating that nSA did not induce significant cytotoxicity in HLE cell at 2000 μg/mL or below. As H2O2 at 250 μM led to a significant cell death, up to 40%, in the HLE cells, this concentration was chosen for the following experiments (). Upon H2O2 stimulation, the effects of nSA on the viability of HLE cells were detected and found that pretreated with nSA (125 to 2000 μg/mL) on HLE cells displayed higher viabilities than that of the non-treated cells (). To quantitatively examine the effect of nSA on H2O2-induced apoptosis in HLE cells, Annexin V/PI double-stained cells were counted using the flow cytometric analysis. As shown in , compared with the control group, the cellular apoptotic rate increased from 3.91 to 39.5% when stimulated with H2O2. However, treatment with 250, 500 and 1000 μg/mL nSA decreased the cell apoptotic rates in a dose-dependent manner to 33.3, 24.5 and 20.1%, respectively. These results suggested that nSA inhibited H2O2-induced apoptosis in HLE cells.
Figure 2. Effects of nSA on cell viability of H2O2-induced cell death in HLE cells. (A) HLE cells were incubated with different concentrations of nSA (62.5–2000 μg/mL) for 24 h. (B) HLE cells were incubated with different concentrations of H2O2 (50–500 μM) for 24 h. (C) HLE cells were preincubated with nSA (62.5–2000 μg/mL) for 2 h before the treatment with 250 μM H2O2 for 24 h. Cell viability was assessed by MTT assay. Data were expressed as mean ± SEM and were obtained from three independent experiments. ###p < 0.001, compared with the untreated control group; *p <0.05, **p <0.01 and ***p <0.001, compared with the H2O2-treated group.
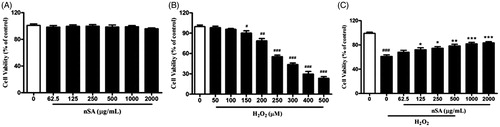
Figure 3. nSA prevented H2O2-induced cellular apoptosis in HLE cells. HLE cells were preincubated with nSA (250, 500 and 1000 μg/mL) for 2 h before the treatment with 250 μM H2O2 for 6 h. The apoptotic cells were stained with Annexin V-FITC and PI, and then detected by flow cytometry. Data were expressed as mean ± SEM and were obtained from three independent experiments. ###p <0.001, compared with the untreated control group; *p <0.05, **p <0.01 and ***p <0.001, compared with the H2O2-treated group.
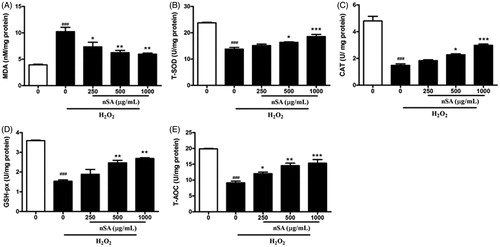
nSA protected HLE cells from H2O2-induced oxidative stress
To examine the antioxidant effects of nSA against H2O2-induced oxidative stress in HLE cells, the content of MDA (a marker for lipid peroxidation), the activities of T-SOD, CAT, GSH-Px, and T-AOC were determined using commercial assay kits. Compared with the control cells, the H2O2-treated cells showed a three-fold increase of MDA content, but decreased activities of T-SOD, CAT, GSH-Px and T-AOC by 1.8–3-folds. In H2O2-treated HLE cells, incubation with nSA (250, 500 and 1000 μg/mL) significantly reduced MDA content in a dose-dependent manner. Importantly, nSA at 1000 μg/mL reduced MDA level by 50%. In addition, incubation with nSA significantly enhanced the activities of T-SOD, CAT, GSH-Px and T-AOC in dose-dependent manners as shown in . These results indicated that nSA possesses potent antioxidant action against H2O2-induced oxidative injury.
Figure 4. nSA inhibited H2O2-induced oxidative stress in HLE cells. HLE cells were preincubated with nSA (250, 500 and 1000 μg/mL) for 2 h before the treatment with 250 μM H2O2 for 24 h. Cells were lysed and then the levels of MDA (A), T-SOD (B), CAT (C), GSH-Px (D) and T-AOC (E) were measured using commercial assay kits. Data were expressed as mean ± SEM and were obtained from three independent experiments. ###p <0.001, compared with the untreated control group; *p <0.05, **p <0.01 and ***p <0.001, compared with the H2O2-treated group.
nSA inhibited H2O2-induced loss of Δψm
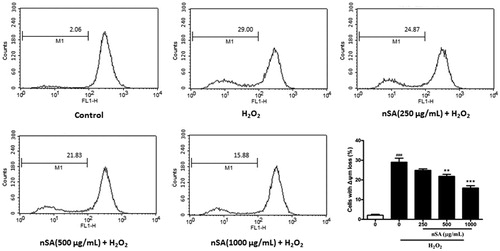
The loss of Δψm indicates impaired mitochondrial function, which is closely associated with the generation of reactive oxygen species (ROS) and cellular apoptosis. As shown in , when exposed to H2O2 (250 μM) for 6 h, approximately 29.0% of the HLE cells showed a significant loss of Δψm. When pretreated with nSA (250, 500 and 1000 μg/mL), merely 24.9, 21.8 and 15.9% of cells showed signs of Δψm loss.
Figure 5. nSA inhibited H2O2-induced dissipation of Δψm in HLE cells. HLE cells were preincubated with nSA (250, 500 and 1000 μg/mL) for 2 h before the treatment with 250 μM H2O2 for 6 h. Cells were incubated with Rh123 (1 μM) for 30 min at 37 °C, and fluorescent intensity were analysed using flow cytometry. Data were expressed as mean ± SEM and were obtained from three independent experiments. #p <0.001, compared with the untreated control group; *p <0.05, **p <0.01 and ***p <0.001, compared with the H2O2-treated group.
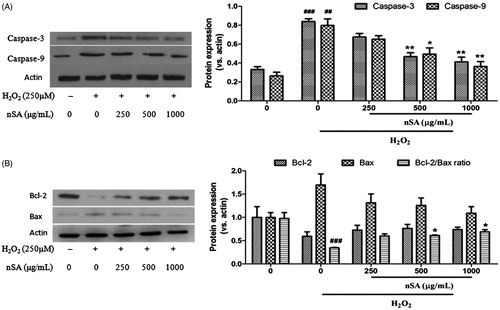
nSA suppressed the activation of caspase-3/-9 and restored the Bcl-2/bax ratio in H2O2-stimulated HLE cells
The effect of nSA on the activation of caspase-3 and caspase-9 in H2O2-treated HLE cells was first determined by means of western blot analysis. As shown in , exposure of HLE cells to H2O2 (250 μM) for 24 h significantly increased the activation of caspase-3 and caspase-9 in HLE cells and pretreatment with nSA significantly inhibited their activation. Subsequently, the expression of Bcl-2 and Bax at the protein levels were also measured. As shown in , compared with the control group, reduced Bcl-2 protein and elevated Bax protein along with a 2.81-fold decrease in the Bcl-2/Bax ratio were detected in HLE cells after H2O2 (250 μM) treatment for 24 h. However, these changes were reversed after pretreatment with nSA. In particular, treatment with 250, 500 and 1000 μg/mL nSA significantly increased the Bcl-2/Bax ratio by 1.73-, 1.76- and 1.98-fold in H2O2-treated HLE cells, respectively.
Figure 6. nSA suppressed the activation of caspase-3 and caspase-9 (A.) and restored the Bcl-2/Bax ratio (B.) in H2O2-induced HLE cells. HLE cells were preincubated with nSA (250, 500 and 1000 μg/mL) for 2 h before the treatment with 250 μM H2O2 for 24 h. Cells were lysed and the expression of caspase-3, caspase-9, Bcl-2 and Bax were evaluated using western blot analysis. Data were expressed as mean ± SEM and were obtained from three independent experiments. ###p <0.001, compared with the untreated control group; *p <0.05 and **p <0.01, compared with the H2O2-treated group.
Discussion
Oxidative damage is thought to play an important role in the pathogenesis of age-related cataracts, and ROS-induced oxidative stress has been recognised as an important mediator of apoptosis in lens epithelial cells (Spector & Garner Citation1981; Truscott Citation2005; Yao et al. Citation2009). H2O2 is a major intracellular ROS and is substantially accumulated in the lens post stimulation or injury (Tang et al. Citation2011). Therefore, in our experiment, H2O2 was used to induce oxidative injury in the lens epithelial cells. In the present work, we found that H2O2-treated cells showed a high level of cellular apoptosis and nSA exhibited a potent effect on inhibiting H2O2-induced cell death in a dose-dependent manner. Our results indicated that nSA may have a protecting effect on oxidative stress-related ocular diseases.
The oxidative stress occurs when the balance of pro-oxidant/antioxidant is disturbed as a result of an increase in free radical production and/or a decrease in radical scavenging capability of antioxidant defence systems (Bai et al. Citation2013). Lipid peroxidation is initiated when abstraction of hydrogen in cellular membranes is accelerated by oxidative stress, and leads to the formation of relatively stable compounds such as MDA. Production of free radicals is blocked by endogenous enzymatic antioxidant systems such as T-SOD, GSH-px and CAT (El-Beltagi & Mohamed Citation2013). In the present study, increased levels of MDA and decreased T-SOD, GSH-px and CAT enzyme activities were observed in HLE cells exposed to H2O2, suggesting that these enzymes were consumed due to increased oxidative stress. After nSA treatment, the content of MDA was significantly decreased in HLE cells, so as the increased activities of SOD, GSH-px and CAT, and T-AOC. Our results indicated that nSA possesses potent antioxidant properties against H2O2-induced oxidative injury.
It is well known that the oxidative stress-induced lens epithelial cell apoptosis plausibly leads to the initiation of cataract formation (Yao et al. Citation2007). Previous studies have shown that mitochondria play a prominent role in the transduction and amplification of the apoptotic response in lens epithelial cells during oxidative stress (Yao et al. Citation2008; Wu et al. Citation2010). According to previous reports, ROS generation might lead to a sustained elevation of [Ca2+], which was associated with mitochondrial dysfunction and a loss of membrane potential (Δψm). Subsequently, the release of mitochondrial protein cytochrome c, a molecule required for the activation of caspases to execute cell death program, was accelerated (Littlewood & Bennett Citation2003). In the present study, H2O2-treated cells showed an increase in the loss of Δψm and the expression levels of caspase-3 and -9 in HLE cells. Our results showed that nSA treatment significantly reduced the loss of Δψm and inhibited the expression of caspase-3 and -9. Thus, we deduced that nSA inhibited H2O2-induced apoptosis via the regulation of mitochondrion-mediated signalling pathway.
The Bcl-2 family members can act as critical regulators of pathways associated with apoptosis, either as inhibitors or as promoters of cell death signals (Burlacu Citation2003). One of the most studied negative regulators of apoptosis is the proto-oncogene Bcl-2. Bcl-2 prevents ROS production and regulates the mitochondrial transitional pore opening by reversing the effect of Bax, thereby blocking the release of cytochrome c and inhibiting caspase activities (Kuwana & Newmeyer Citation2003). An altered ratio of anti-apoptotic/pro-apoptotic Bcl-2 family proteins is critical in determining whether apoptosis assumes (Gross et al. Citation1999). Consistent with aforementioned results, Bcl-2/Bax ratio was significantly decreased in HLE cells when exposed to H2O2 and was reversed in the presence of nSA. These results suggest that nSA could protect HLE cells from oxidative stress through modulating the Bcl-2/Bax ratio.
Conclusions
In conclusion, results from this study indicated that nSA exerted a potent effect on protecting HLE cells from oxidative stress through modulating radical scavenging capability, mitochondrial membrane potential, caspases and Bcl-2/Bax. Our findings suggest a potential implication of nSA in the prevention of cataractogeneis.
Funding information
This research was supported by the National Natural Science Foundation of China (No. 21102025) and a grant from The Guizhou Science and Technology Department (ZY[2011]3006).
Acknowledgements
The authors would like to thank Dr. Siu-Wai Tsang (School of Chinese Medicine, Hong Kong Baptist University) for her critical review of this manuscript.
Disclosure statement
The authors report that they have no conflicts of interest.
References
- Bai J, Dong L, Song Z, Ge H, Cai X, Wang G, Liu P. 2013. The role of melatonin as an antioxidant in human lens epithelial cells. Free Radic Res. 47:635–642.
- Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 72:248–254.
- Burlacu A. 2003. Regulation of apoptosis by Bcl-2 family proteins. J Cell Mol Med. 7:249–257.
- El-Beltagi HS, Mohamed H. 2013. Reactive oxygen species, lipid peroxidation and antioxidative defense mechanism. Not Bot Horti Agrobo. 41:44–57.
- Giblin FJ. 2000. Glutathione: a vital lens antioxidant. J Ocul Pharmacol Ther. 16:121–135.
- Gross A, Mcdonnell JM, Korsmeyer SJ. 1999. Bcl-2 family members and the mitochondria in apoptosis. Genes Dev. 13:1899–1911.
- Guiyang HBO, editor. 1959. Guiyang folk medicinal herbs. Guiyang: Guiyang People's Publishing House.
- Han X, Zhu S, Wang B, Chen L, Li R, Yao W, Qu Z. 2014. Antioxidant action of 7,8-dihydroxyflavone protects PC12 cells against 6-hydroxydopamine-induced cytotoxicity. Neurochem Int. 64:18–23.
- Hou F, Tang L, Wang J, Xiao H, Guo Z. 2012. Semiaquilegia root extract, preparation method and application thereof. Chinese Patent CN101773553B.
- Kuwana T, Newmeyer DD. 2003. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol. 15:691–699.
- Li L, Duker JS, Yoshida Y, Niki E, Rasmussen H, Russell RM, Yeum KJ. 2009. Oxidative stress and antioxidant status in older adults with early cataract. Eye (Lond.) 23:1464–1468.
- Littlewood TD, Bennett MR. 2003. Apoptotic cell death in atherosclerosis. Curr Opin Lipidol. 14:469–475.
- Meekins LC, Afshari NA. 2012. Ever-evolving technological advances in cataract surgery: can perfection be achieved? Curr Opin Ophthalmol. 23:1–2.
- Mok JW, Chang DJ, Joo CK. 2014. Antiapoptotic effects of anthocyanin from the seed coat of black soybean against oxidative damage of human lens epithelial cell induced by H2O2. Curr Eye Res. 39:1090–1098.
- Niu F, Chang HT, Jiang Y, Cui Z, Chen FK, Yuan JZ, Tu PF. 2006. New diterpenoids from Semiaquilegia adoxoides. J Asian Nat Prod Res. 8:87–91.
- Ou Y, Liao G, Yuan Z, Wu W. 2012. Protective effect of recombinant hirudin variant III against galactose-mediated rat lens epithelial cell damage. Curr Eye Res. 37:187–194.
- Paron I, D'elia A, D'ambrosio C, Scaloni A, D'Aurizio F, Prescott A, Damante G, Tell G. 2004. A proteomic approach to identify early molecular targets of oxidative stress in human epithelial lens cells. Biochem J. 378:929–937.
- Salm M, Belsky D, Sloan FA. 2006. Trends in cost of major eye diseases to Medicare, 1991 to 2000. Am J Ophthalmol. 142:976–982.
- Spector A, Garner WH. 1981. Hydrogen peroxide and human cataract. Exp Eye Res. 33:673–681.
- Su Y, Zhang Z, Guo C. 2004. A new nitroethylphenolic glycoside from Semiaquilegia adoxoides. Fitoterapia 75:420–422.
- Tang X, Yao K, Zhang L, Yang Y, Yao H. 2011. Honokiol inhibits H(2)O(2)-induced apoptosis in human lens epithelial cells via inhibition of the mitogen-activated protein kinase and Akt pathways. Eur J Pharmacol. 650:72–78.
- Truscott RJW. 2005. Age-related nuclear cataract-oxidation is the key. Exp Eye Res. 80:709–725.
- Wu H, Xing K, Lou MF. 2010. Glutaredoxin 2 prevents H(2)O(2)-induced cell apoptosis by protecting complex I activity in the mitochondria. Biochim Biophys Acta. 1797:1705–1715.
- Xiao HT, Qi XL, Liang Y, Lin CY, Wang X, Guan ZZ, Hao XY. 2014. Membrane permeability-guided identification of neuroprotective components from Polygonum cuspidatun. Pharm Biol. 52:356–361.
- Yao H, Tang X, Shao X, Feng L, Wu N, Yao K. 2007. Parthenolide protects human lens epithelial cells from oxidative stress-induced apoptosis via inhibition of activation of caspase-3 and caspase-9. Cell Res. 17:565–571.
- Yao K, Ye P, Zhang L, Tan J, Tang X, Zhang Y. 2008. Epigallocatechin gallate protects against oxidative stress-induced mitochondria-dependent apoptosis in human lens epithelial cells. Mol Vis. 14:217–223.
- Yao K, Zhang L, Ye PP, Tang XJ, Zhang YD. 2009. Protective effect of magnolol against hydrogen peroxide-induced oxidative stress in human lens epithelial cells. Am J Chin Med. 37:785–796.
- Zheng Y, Liu Y, Ge J, Wang X, Liu L, Bu Z, Liu P. 2010. Resveratrol protects human lens epithelial cells against H2O2-induced oxidative stress by increasing catalase, SOD-1, and HO-1 expression. Mol Vis. 16:1467–1474.