
Abstract
Context Mangiferin has been reported to possess a potential hypouricaemic effect. However, the pharmacokinetic studies in rats showed that its oral bioavailability was only 1.2%, suggesting that mangiferin metabolites might exert the action.
Objective The hypouricaemic effect and the xanthine oxidase inhibition of mangiferin and norathyriol, a mangiferin metabolite, were investigated. Inhibition of norathyriol analogues (compounds 3–9) toward xanthine oxidase was also evaluated.
Materials and methods For a dose-dependent study, mangiferin (1.5–6.0 mg/kg) and norathyriol (0.92–3.7 mg/kg) were administered intragastrically to mice twice daily for five times. For a time-course study, mice received mangiferin and norathyriol both at a single dose of 7.1 μmol/kg. In vitro, inhibition of test compounds (2.4–2.4 mM) against xanthine oxidase activity was evaluated by the spectrophotometrical method. The inhibition type was identified from Lineweaver–Burk plots.
Results Norathyriol (0.92, 1.85 and 3.7 mg/kg) dose dependently decreased the serum urate levels by 27.0, 33.6 and 37.4%, respectively. The action was more potent than that of mangiferin at the low dose, but was equivalent at the higher doses. Additionally, the hypouricaemic action of them exhibited a time dependence. In vitro, norathyriol markedly inhibited the xanthine oxidase activities, with the IC50 value of 44.6 μM, but mangiferin did not. The kinetic studies showed that norathyriol was an uncompetitive inhibitor by Lineweaver–Burk plots. The structure–activity relationships exhibited that three hydroxyl groups in norathyriol at the C-1, C-3 and C-6 positions were essential for maintaining xanthine oxidase inhibition.
Discussion and conclusion Norathyriol was responsible for the hypouricaemic effect of mangiferin via inhibiting xanthine oxidase activity.
Introduction
Uric acid is produced by the oxidation of hypoxanthine and xanthine by xanthine oxidase. In humans, uric acid is the end product of purine catabolism, whereas in other mammals, it is further degraded into allantoin by uricase. The gene-encoding uricase underwent mutational silencing during hominid evolution (Oda et al. Citation2002). The consequence of uricase inactivation is the appearance of urate levels that are much higher in humans in comparison with other mammals. Documented evidence shows that hyperuricaemia is associated with not only gout but also cardiovascular disorder, nephrolithiasis, renal dysfunction and diabetes (Edwards Citation2008; Puig & Martínez Citation2008; Karis et al. Citation2014). The control of the key factor, uric acid, might be beneficial for the prevention and treatment of these diseases. A useful therapeutic approach for the management of patients with hyperuricaemia and gout is inhibition of xanthine oxidase activity with xanthine oxidase inhibitors, such as allopurinol and febuxostat.
Mangiferin (, compound 1), 1,3,6,7-tetrahydroxyxanthone-C-2-β-d-glucoside, is a natural glucosyl xanthone from the leaves of Mangifera indica L. (Anacardiaceae). In the previous study, we reported that mangiferin had potential hypouricaemic effects. Given intragastrically at a 1.5–6.0 mg/kg dose, mangiferin significantly decreased the serum urate levels in hyperuricaemic mice induced by potassium oxonate (an uricase inhibitor), in a dose-dependent manner (Li & Chen Citation2010; Niu et al. Citation2012). The potency of hypouricaemic action was similar to that of allopurinol. However, the pharmacokinetic studies in rats showed that the oral bioavailability of mangiferin was only 1.2%, indicating poor oral absorption (Wang et al. Citation2007; Han et al. Citation2010). Judged from the poor bioavailability, it is unbelievable that mangiferin has such a strong hypouricaemic effect. So, we may assume that this effect should not come from mangiferin itself but its metabolites.
Table 1. Chemical structures of mangiferin and its analogues.
Norathyriol (, compound 2), the aglycone of mangiferin, is a principal metabolite of mangiferin in the intestinal tract. It has been shown that mangiferin was deglycosylated into norathyriol by intestinal bacterium after oral administration (Sanugul et al. Citation2005; Liu et al. Citation2011). Mangiferin induced Bacteroides species MANG, a specific intestinal bacterium, to produce a C-glucosyl-cleavage enzyme that transformed mangiferin to norathyriol by cleaving the C–C bond of mangiferin, and norathyriol was detected in urine in rats given oral mangiferin (Sanugul et al. Citation2005; Wang et al. Citation2007), suggesting that mangiferin was absorbed in a form of norathyriol. Thus, we hypothesise that norathriol is responsible for the observed hypouricaemic effect. In order to verify the possibility, the hypouricaemic effect of norathyriol was compared with mangiferin in the hyperuricaemic mice induced by potassium oxonate, in the present paper. In addition, the mechanism of mangiferin-mediated hypouricaemic action has not been elucidated. Herein, the inhibitory effects of mangiferin and its aglycone, norathyriol, on the xanthine oxidase activity, were investigated in vitro. Other xanthone compounds (, compounds 3–9) were also evaluated, aiming at looking for possible existing structure–activity relationship .
Materials and methods
Animals
Adult Kunming mice weighing 18–22 g were obtained from the Experimental Animal Center, Kunming Medical University (Certificate no. SCXK 2005-0008). The animals were housed on a constant 12 h light/dark cycle in a temperature- and humidity-controlled room, and allowed free access to solid food and tap water for 1 week prior to the experiments. All the procedures were performed in accordance with the Institute Ethical Committee for Experimental Animal Use (GB14925-2001).
Reagents
Xanthine oxidase (EC 1.1.3.22) from bovine milk (0.4 units/mg protein), xanthine, sodium pyrophosphate, potassium oxonate and allopurinol were purchased from Sigma Aldrich Chemical Co. (St. Louis, MO). The biochemical kits used in the experiments were the products of Nanjing Jiancheng Bioengineering Institute. All other reagents were commercially available and of analytical grade.
Preparation of compounds tested
Mangifera indica L. (Anacardiaceae) leaves were collected from Baise of Guangxi Province, China, in October 2012 and identified by Prof. Haiyang Liu, Kunming Institute of Botany, the Chinese Academy of Science, where a voucher specimen (No. 121016) has been deposited. Mangiferin (1, purity > 90% by HPLC) was isolated from M. indica leaves according to a method described previously (Ge et al. Citation2011). Briefly, the EtOH extract of M. indica leaves was partitioned between EtOAc and water to afford an EtOAc-soluble portion. The water fraction was then passed through the macroporous resin and eluted successively with H2O and 95% EtOH. The 95% EtOH fraction was subjected to subsequent silica gel, Sephadex LH-20 and Rp-18 silica gel column chromatography to yield mangiferin (1). Norathyriol synthesis (2, purity > 98% by HPLC) was performed according to the method described previously (Zhou et al. Citation2011; Li et al. Citation2012). In brief, norathyriol was synthesised from 2-bromo-4,5-dimethoxybenzoic acid and trimethoxybenzene for a total yield of 51% through four steps including Friedel–Crafts acetylation, selective demethylation, cyclisation via the Ullmann reaction and overall demethylation. The other compounds (3–9) were isolated from Polygala crotalarioides Buch.-Ham. (Polygalaceae), prepared as presented in the previous study (Deng et al. Citation2006; Hua et al. Citation2006, Citation2007). All the compounds were dissolved in 0.3% sodium pyrophosphate buffer with 0.15% polyethylene glycol 400 (PEG-400) for a zymologic assay and in 0.5% sodium carboxymethyl cellulose (CMC-Na) for in vivo study.
Hypouricaemic effects of compounds 1 and 2 in potassium oxonate-induced hyperuricaemic mice
The hyperuricaemic mice were induced by an intraperitoneal injection with uricase inhibitor potassium oxonate (Stavric et al. Citation1975; Hall et al. Citation1990). Male mice were divided into the normal control group, the untreated hyperuricaemic control group and treated hyperuricaemic groups. The normal and hyperuricaemic control mice were treated with vehicles, whereas the other hyperuricaemic mice received compounds 1 and 2 and allopurinol dissolved in 0.5% sodium carboxymethyl cellulose (CMC-Na) or PEG-400. For a dose-dependent study, compound 1 (1.5, 3.0 and 6.0 mg/kg) and compound 2 (0.92, 1.85 and 3.7 mg/kg) were administered intragastrically to mice twice daily at 9:00 a.m. and 4:00 p.m. for 2.5 d, respectively. The last dose was given at 1 h after intraperitoneal injection with potassium oxonate (500 mg/kg) to increase serum uric acid levels. One hour after administration of the last dose, blood samples were collected from the mice via the orbital vein under ether anaesthesia. Serum uric acid levels were determined using the phosphotungstic acid method (Carroll et al. Citation1971). For the time-course study, the hyperuricaemic mice received compound 1 or compound 2 both at a single dose of 7.1 μmol/kg by intragastrical or intraperitoneal administration. Serum uric acid levels were determined at 30, 60 and 90 min after administration. Food was withdrawn from the animals 2 h prior to blood sample collection.
Xanthine oxidase activity assay
The enzymatic activity was measured spectrophotometrically by continuously measuring uric acid formation at 295 nm with xanthine as a substrate (Kong et al. Citation2000; Niu et al. Citation2011). The reaction mixture contained 0.3% sodium pyrophosphate buffer (pH 8.3) and 8.35 U/L xanthine oxidase with or without the test compounds. Allopurinol was used as a positive control. After preincubation at 25 °C for 15 min, uric acid formation in the reaction mixture was initiated by 400 μM xanthine addition. In the enzyme kinetics tests, the xanthine concentrations used were varied from 10 to 1200 μM. The xanthine oxidase inhibition type that was mediated by the test compounds was identified from Lineweaver–Burk plots, and the Ki value was analysed using GraphPad Prism 5.0 software (GraphPad Software Inc., La Jolla, CA).
Statistical analysis
All the data were expressed as the mean ± SD. IC50 values were determined from the concentration-dependent curves. Data from the in vivo experiments were subjected to one-way analysis of variance followed by Dunnett’s multiple comparison tests using GraphPad Prism version 5.0 software (GraphPad Software Inc., La Jolla, CA). Statistical significance was accepted at p < 0.05.
Results
Effects of compounds 1 and 2 on serum uric acid levels in potassium oxonate-treated mice
As reported in , an intraperitoneal potassium oxonate injection markedly increased the serum uric acid levels of mice, compared with normal mice. When the hyperuricaemic mice received compound 2 at a dose of 0.92, 1.85 and 3.70 mg/kg (3.5, 7.1 and 14.2 μmol/kg) twice daily for 2.5 d, respectively, the serum uric acid levels were significantly attenuated in the hyperuricaemic mice, compared with the untreated hyperuricaemic mice (p < 0.05). The serum uric acid levels were decreased by 27.0%, 33.6% and 37.4%. Similar effects were obtained in the compound 1-treated groups, with the decrease of the serum uric acid by 19.5, 29.5 and 34.4%, respectively, at the doses of 1.5, 3.0 and 6.0 mg/kg (3.5, 7.1 and 14.2 μmol/kg). Moreover, compound 2 at the low dose was stronger in potency than that of compound 1 (p < 0.05), and weaker than that of allopurinol (p < 0.05) ().
Figure 1. Effects of intragastric administration of compounds 1 and 2 on serum uric acid levels in uricase inhibitor potassium oxonate-treated mice. CON, normal control; M, hyperuricaemic control. The data are expressed as the means ± SD for 10 mice per group. ap < 0.05 or less versus the control group; bp < 0.05 or less versus the hyperuricaemic group; cp < 0.05 or less versus the compound 1 (1.5 mg/kg) treated group; dp < 0.05 or less versus the compound 2 (0.93 mg/kg) treated group; ep < 0.05 or less versus the allopurinol-treated group (Dunnett’s test).
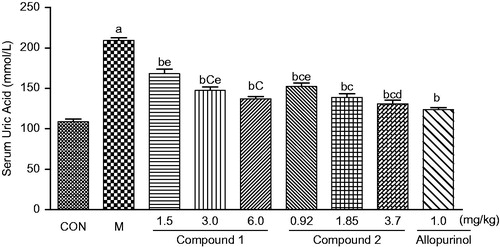
illustrated that the serum uric acid levels in the hyperuricaemic mice were decreased after administration of compound 1 or 2 in a time-dependent manner. Thirty minutes after a single intragastric dose (7.1 μmol/kg), both compounds 1 and 2 significantly reduced the serum uric acid levels, when compared with the untreated hyperuricaemic mice (p < 0.05). After intravenous administration of compounds 2 and 1 at the same molar dose, the hypouricaemic action was detected after 30 and 60 min, respectively, suggesting that compound 2 functioned earlier than compound 1.
Table 2. Effects of compounds 1 and 2 on serum uric acid levels in the hyperuricaemic mice induced by uricase inhibitor potassium oxonate: a time-course study.
The xanthine oxidase inhibitory activities of compounds 1–9
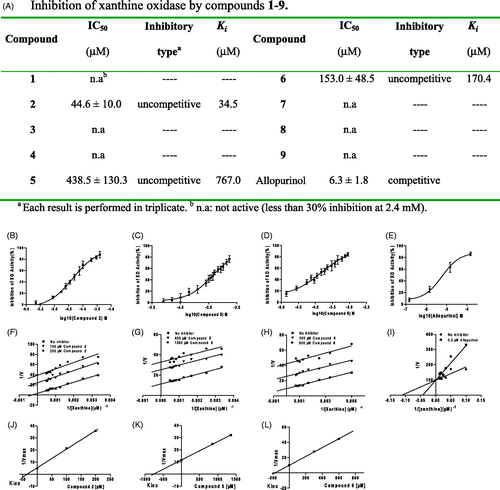
The xanthine oxidase inhibitory activities of compounds 1–9 are described in . From these data, it can be concluded that compounds 1, 3, 4, 7, 8 and 9 did not demonstrate any inhibition even at concentrations up to 2.4 mM. However, compounds 2, 5 and 6 significantly inhibited the xanthine oxidase activities in a concentration-dependent manner. The potency of inhibition was weaker than that of allopurinol. The IC50 values were 44.6, 483.5 and 153 μM for compounds 2, 5 and 6, respectively, and 6.3 μM for allopurinol. Lineweaver–Burk double reciprocal plots showed that compounds 2, 5 and 6 were uncompetitive xanthine oxidase inhibitors, and the inhibition constant Ki values were 34.5, 767.0 and 170.4 μM, respectively. The action mode of them was different from allopurinol, a competitive inhibitor.
Figure 2. Inhibition of xanthine oxidase by compounds 1–9. (A) IC50, inhibitory type and Kies values of test compounds. (B–E) Concentration-dependent xanthine oxidase (XO) inhibition by compounds 2, 5, 6 and allopurinol. Data presented are the mean values ± SD. for eight independent experiments. (F–L) Inhibition kinetics of xanthine oxidase by compounds 2, 5, 6 and allopurinol. Each point is the average value from three independent experiments.
Discussion
In the previous study, we reported that compound 1 at a dose of 1.5 mg/kg markedly decreased the serum uric acid levels in the hyperuricaemic mice induced by uricase inhibitor potassium oxonate in a manner that was similar to allopurinol treatment (Li & Chen Citation2010; Niu et al. Citation2012), although the oral bioavailability of compound 1 was only 1.2% (Han et al. Citation2010). This hinted that the observed hypouricaemic effect of compound 1 was related to its metabolites. In view of the fact that compound 2 is a primary metabolite of compound 1 (Sanugul et al. Citation2005; Wang et al. Citation2007), in the current study, we investigated the hypouricaemic effect of compound 2 and compared with compound 1 in potency. The results showed that compound 2 significantly improved the hyperuricaemia induced by potassium oxonate in mice, exhibiting the hypouricaemic effect in a dose-dependent manner. Similar effects were obtained in the compound 1-treated groups. Notably, the serum uric acid levels were decreased by 27.0% for compound 2 at the dose of 0.93 mg/kg and by 19.5% for compound 1 at the dose of 1.5 mg/kg, exhibiting that compound 2 was more potent than compound 1. It was suggested that the required dose for compound 1 was greater than the aglycone form – compound 2 – in the lower dose group (3.5 μmol/kg) from , whereas not in higher dose groups (7.1 and 14.2 μmol/kg). The phenomenon implied that the more compound 2 was transformed from compound 1 at the higher dose. On the other hand, the time-course study showed that the onset of the hypouricaemic action for both compounds 1 and 2 was at 30 min after intragastric administration. It could be inferred that compound 1 could be transformed quickly to compound 2 in intestinal tract, which was consistent with the outcomes reported by Sanugul et al. (Citation2005). Subsequently, compound 2 might be absorbed to perform the hypouricaemic action. These findings suggested that the hypouricaemic effect of compound 1 might be related to compound 2. This is the first time to demonstrate that compound 2 might be an important active metabolite of compound 1 for the hypouricaemic action.
In addition, we have drawn attention to the fact that compound 1 also decreased the serum uric acid levels in the hyperuricaemic mice after intravenous injection, although the hypouricaemic action of compound 1 was transitory and the onset was slower than that of compound 2. It could be inferred that compound 1 might be metabolised in liver besides gastrointestinal tract, and compound 2 was not the only active metabolite of compound 1. As reported by Wang et al. (Citation2007) and Liu et al. (Citation2011), compound 1 could be transformed to many metabolites in vivo. These metabolites might be also associated with observed hypouricaemic action. Further research need to be done for more evidence.
Xanthine oxidase (EC 1.1.3.22), a key enzyme in uric acid production, is a molybdenum hydroxylase superfamily member and complex flavoprotein that is composed of two identical subunits measuring approximately 145 kDa. Each catalytically independent subunit contains molybdopterin (85 kDa) with its four redox centres aligned in an almost linear fashion, a moiety containing two non-identical Fe2S2 iron sulphur centres (20 kDa) and one flavin adenine dinucleotide (FAD) (40 kDa) (Borges et al. Citation2002). Xanthine oxidase catalyses oxidative hydroxylation of purine substrates at the molybdenum centre to form uric acid.
To clarify the mechanism of compound 1 for hyperuricaemia therapy, the effects of compounds 1 and 2 against the xanthine oxidase activity were investigated in vitro. Interestingly, compound 1 did not suppress the enzymatic activity even at concentrations up to 2.4 mM. In contrast, compound 2, the aglycone of compound 1, significantly inhibited the activities of xanthine oxidase in a concentration-dependent manner, with an IC50 value of 44.6 μM. This finding was consistent with the results obtained in vivo, further suggesting that the hypouricaemic action of compound 1 might be related to its metabolite compound 2. In other words, the hypouricaemic action of compound 1 was attributed to compound 2-mediated xanthine oxidase inhibition.
The xanthine oxidase-catalysed hydroxylation is initiated by the abstraction of a proton from the Mo–OH group by an active site glutamate residue that is universally conserved in the family of molybdenum-containing enzymes. From the molybdenum centre, electrons are passed sequentially via two [2Fe–2S] clusters to an FAD site where they are finally passed to NAD+ or O2 (Pauff & Hille Citation2009). A competitive inhibitor such as allopurinol is structurally related to the substrate and competes with the substrate for the active site. Allopurinol is hydroxylated by xanthine oxidase to alloxanthine (oxypurinol), which coordinates tightly to the reduced form of the molybdenum centre specifically and replaces the native enzyme Mo–OH group. Febuxostat is a mixed-type xanthine oxidase inhibitor (Okamoto et al. Citation2003; Takanoa et al. Citation2005) that can bind both the free enzyme and the enzyme–substrate complex because of structural dissimilarity to the substrate. In the current study, the xanthine oxidation inhibition kinetics demonstrated that compound 2 was an uncompetitive inhibitor by Lineweaver–Burk plots, suggesting that compound 2 only bound the xanthine oxidase–xanthine complex to form an enzyme–substrate–inhibitor complex, thus suppressing uric acid production. This mode of inhibition was much different from that of allopurinol and febuxostat even though they each target xanthine oxidase, which might be beneficial for hyperuricaemia therapy.
Moreover, to research structure–activity relationships, the xanthine oxidase inhibition was also investigated using seven compound 1 aglycone analogues (compounds 3–9) in vitro. The results showed that only two of the seven analogues significantly inhibited the activities of xanthine oxidase. The mode of inhibition was uncompetitive as assessed by Lineweaver–Burk plots, which was similar to compound 2, but was not as potent. Introducing an OMe group at C-2 or OMe groups at C-2, C-7 and C-8 in compound 2 might dramatically reduce its xanthine oxidase inhibition, whereas the xanthine oxidase inhibition vanished while the OH-1 or OH-3 was replaced with an OMe group, and the OH-6 or OH-7 was substituted with a hydrogen atom or OMe group. These data implied that the hydroxy groups present at the C-2, C-3 and C-6 positions of compound 2 were necessary for maintaining xanthine oxidase inhibition. Moreover, the biological activity of compound 2 disappeared after introducing a glucosyl group at the C-2 position. Similarly, flavonol glycosides such as quercetin and kaempferol glycosides that had glycoside substitutions at the C7 and C3 hydroxyl groups exhibited much weaker inhibitory activities compared with their aglycone forms (Borges et al. Citation2002; Spanou et al. Citation2012). Similar results occurred in luteolin and apigenin glycosides (Flemmig et al. Citation2011).
These findings suggested that the presence of the glycosides might interfere with inhibitor–enzyme binding. The structure–activity relationships of xanthone derivatives displayed no significant influence on xanthine oxidase inhibition after introducing electron donor or electron withdrawing substituents on the benzyl group. Compounds with a cyano group at the para position demonstrated significant xanthine oxidase inhibition, which was followed by compounds with a chloride group at the benzyl para position, with a methyl group at the para position, with a chloride group at the ortho position and with a methyl group at the ortho position, respectively, which suggested that para-substituted benzyl groups are of greater benefit to the inhibition than compounds with the same substitutes at the ortho or meta positions (Hu et al. Citation2011).
Conclusions
Compound 2 might be an important active metabolite of compound 1. The hypouricaemic action of compound 1 might result from norathyriol via xanthine oxidase inhibition.
Funding information
This work was supported by National Natural Science Foundation of China (30960453 and 81360505), Yunnan Provincial Science Foundation (2008IF009, 2013FA017 and 2010CD224), and Yunnan Provincial academician workstation of H. F. Kung.
Disclosure statement
The authors report that they have no conflicts of interest.
References
- Borges F, Fernandes E, Roleira F. 2002. Progress towards the discovery of xanthine oxidase inhibitors. Curr Med Chem. 9:195–217.
- Carroll JJ, Coburn J, Douglas R, Babson AL. 1971. A simplified alkaline phosphotungstate assay for uric acid in serum. Clin Chem. 17:158–160.
- Deng SM, Yang XH, Zhao YX, Zhou J. 2006. New xanthones from Polygala crotalarioides. Chem Res Chinese Univ. 22:400–402.
- Edwards NL. 2008. The role of hyperuricemia and gout in kidney and cardiovascular disease. Clev Clin J Med. 75 (Suppl 5):S13–S16.
- Flemmig J, Kuchta K, Arnhold J, Rauwald HW. 2011. Olea europaea leaf (Ph.Eur.) extract as well as several of its isolated phenolics inhibit the gout-related enzyme xanthine oxidase. Phytomedicine. 18:561–566.
- Ge DD, Zhang Y, Liu EW, Wang T, Hu LM. 2011. Chemical constituents of Mangifera indica leaves (I). Chin Tradit Herb Drugs. 42:428–431.
- Hall IH, Scoville JP, Reynolds DJ. Simlot R, Duncan P. 1990. Substituted cyclic imides as potential anti-gout agents. Life Sci. 46:1923–1927.
- Han DD, Chen CJ, Zhang C, Zhang Y, Tang X. 2010. Determination of mangiferin in rat plasma by liquid-liquid extraction with UPLC-MS/MS. J Pharm Biomed Anal. 51:260–263.
- Hu L, Hu HG, Wu WF, Chai XY, Luo JF, Wu Q. 2011. Discovery of novel xanthone derivatives as xanthine oxidase inhibitors. Bioorg Med Chem Lett. 21:4013–4015.
- Hua Y, Chen CX, Liu YQ, Chen SK, Zhou J. 2006. Three new xanthones from Polygala crotalarioides. Chin Chem Lett. 17:773–775.
- Hua Y, Chen CX, Liu YQ, Zhou J. 2007. Two new xanthones from Polygala crotalarioides. J Asian Nat Prod Res. 9:73–75.
- Karis E, Crittenden DB, Pillinger MH. 2014. Hyperuricemia, gout, and related comorbidities: cause and effect on a two-way street. South Med J. 107:235–241.
- Kong LD, Zhang Y, Pan X, Tan RX, Cheng CHK. 2000. Inhibition of xanthine oxidase by liquiritigenin and isoliquiritigenin isolated from Sinofranchetia chinensis. Cell Mol Life Sci. 57:500–505.
- Li L, Chen ZH. 2010. New application of mangiferin. CN101214254B.
- Li L, Song LD, Liu X, Gao S. 2012. The method of total synthesis of norathyriol. CN102002031B.
- Liu HH, Wang K, Tang YH, Sun ZL, Jian LH, Li ZX, Wu B, Huang CG. 2011. Structure elucidation of in vivo and in vitro metabolites of mangiferin. J Pharm Biomed Anal. 55:1075–1082.
- Niu Y, Lu W, Gao L, Lin H, Liu X, Li L. 2012. Reducing effect of mangiferin on serum uric acid levels in mice. Pharm Biol. 50:1177–1182.
- Niu Y, Zhu H, Liu J, Fan H, Sun L, Lu W, Liu X, Li L. 2011. 3,5,2′,4′-Tetrahydroxychalcone, a new non-purine xanthine oxidase inhibitor. Chem Biol Interact. 189:161–166.
- Oda M, Satta Y, Takenaka O, Takahata N. 2002. Loss of urate oxidase activity in hominoids and its evolutionary implications. Mol Biol Evol. 19:640–653.
- Okamoto K, Satta Y, Takenaka O, Takahata N. 2003. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme–inhibitor complex and mechanism of inhibition. J Biol Chem. 278:1848–1855.
- Pauff JM, Hille R. 2009. Inhibition studies of bovine xanthine oxidase by luteolin, silibinin, quercetin, and curcumin. J Nat Prod. 72:725–731.
- Puig JG, Martínez MA. 2008. Hyperuricemia, gout and the metabolic syndrome. Curr Opin Rheumatol. 20:187–191.
- Sanugul K, Akao T, Li Y, Kakiuchi N, Nakamura N, Hattori M, et al. 2005. Isolation of a human intestinal bacterium that transforms mangiferin to norathyriol and inducibility of the enzyme that cleaves a C-glucosyl bond. Biol Pharm Bull. 28:1672–1678.
- Spanou C, Veskoukis AS, Kerasioti T, Kontou M, Angelis A, Aligiannis N, Skaltsounis AL, Kouretas D. 2012. Flavonoid glycosides isolated from unique legume plant extracts as novel inhibitors of xanthine oxidase. PLoS One 7:e32214.
- Stavric B, Clayman S, Gradd RE, Hebert D. 1975. Some in vivo effects in the rat induced by chlorprothixene and potassium oxonate. Pharmacol Res Commun. 7:117–124.
- Takanoa Y, Hase-Aoki K, Horiuchi H, Zhao L, Kasahara Y, Kondo S, Becker MA. 2005. Selectivity of febuxostat, a novel non-purine inhibitor of xanthine oxidase/xanthine dehydrogenase. Life Sci. 76:1835–1847.
- Wang H, Ye G, Ma CH, Tang YH, Fan MS, Li ZX, Huang CG. 2007. Identification and determination of four metabolites of mangiferin in rat urine. J Pharm Biomed Anal. 45:793–798.
- Zhou RG, Yang ZX, Yang J, Wang J, Yang B, Zhang W. 2011. Total synthesis of norathyriol. App Chem Ind. 40:1516–1518.