
Abstract
Context Simvastatin (SV) and bergamottin (BGM) are known to exhibit diverse anti-cancer and anti-inflammatory activities.
Objective Very little is known about the potential efficacy of combination of these two agents to potentiate TNF-induced apoptosis in human chronic myelogenous leukaemia (CML).
Materials and methods In the present study, we investigated whether SV combined with BGM mediates its effect through suppression of NF-κB-signalling pathway.
Results We found that the combination treatment enhanced cytotoxicity and potentiated the apoptosis induced by TNF as indicated by intracellular esterase activity, Annexin V staining and caspase activation. This effect of co-treatment correlated with down-regulation of various gene products that mediate cell proliferation (cyclin D1), cell survival (cIAP-1, Bcl-2, Bcl-xL and Survivin), invasion (MMP-9) and angiogenesis (VEGF); all known to be regulated by NF-κB. SV combined with BGM also produced TNF-induced cell-cycle arrest in S-phase and this arrest correlated with a concomitant increase in the levels of cyclin-dependent inhibitor p21 and p27. The combination therapy inhibited TNF-induced NF-κB activation, IκBα degradation and p65 translocation to the nucleus as compared with the treatment with individual agents alone. Besides, SV combined with BGM did not significantly potentiate apoptotic effect induced by TNF in p65−/− cells, as compared with wild-type fibroblasts.
Discussion and conclusion Our results provide novel insight into the role of SV and BGM in potentially preventing and treating cancer through modulation of NF-κB signalling pathway and its regulated gene products.
Keywords:
Introduction
Grapefruit juice has been found to significantly increase oral bioavailability of several drugs metabolized by cytochrome P450 3A4 (P450 3A4) through inhibiting the enzymatic activity and decreasing the content of intestinal P450 3A4 (He et al. Citation1998). Bergamottin (BGM) is a member of the furanocoumarin family and is most commonly found in grapefruit juice (Paine et al. Citation2005). BGM is also a known drug inhibitor of cytochrome P450 34A (Bailey et al. Citation1998). For this reason, it is often recommended that patients avoid consumption of grapefruit juice if currently prescribed medication such as statins, anti-histamines and drugs in several other classes. There are many drugs that can be metabolized by the cytochrome P450 34A enzyme, thus the inhibition of this enzyme could be a cause for deep concern, although the results from its suppression are not always considered to have adverse effects (Girennavar et al. Citation2007). BGM has been shown to be a mixed-type inhibitor of simvastatin (SV) metabolism in human and rat microsomes and in rat hepatocytes (Le Goff-Klein et al. Citation2003).
Recently, SV has been studied for its potential use in cancer therapy. Several in vitro studies have shown that SV displays anticancer activity, but at concentrations unlikely to be achieved in patients receiving typical anti-hyperlipidemic treatment doses. Thus, several clinical trials were conducted to study the tolerability of high-dose statins in cancer patients. The maximum-tolerated dose of SV has been determined to be 15 mg/kg/d, 25-fold higher than a typical dose (Ahmed Citation2013). As such high concentrations are not compatible to be used in cancer patients, and thereby preclude use of SV as a single agent for cancer therapy. Besides, the co-administration of statins with other drugs that can interfere with its metabolism through inhibition of CYP450 system may further potentiates the major adverse effects of statins including hepatotoxicity and myotoxicity (Chan et al. Citation2003). As a consequence of these limitations to use of SV particularly at high doses, alternative strategies to use lower dosage of SV with high bioavailability are urgently needed.
Apoptosis, invasion, angiogenesis and inflammation have been shown to be regulated by the nuclear transcription factor NF-κB (Aggarwal Citation2004). NF-κB is a family of Rel domain-containing proteins present in the cytoplasm of all cells, where they are kept in an inactive state by a family of anchorin domain-containing proteins that includes IκBα, IκBβ, IκBγ, IκBɛ, bcl-3, p105 and p100. Under resting conditions, NF-κB consists of a heterotrimer of p50, p65 and IκBα in the cytoplasm (Aggarwal Citation2003); only when activated and translocated to the nucleus is the sequence of events leading to activation initiated. The activation of NF-κB involves the phosphorylation, ubiquitination and degradation of IκBα and phosphorylation of p65, which in turn lead to the translocation of NF-κB to the nucleus where it binds to specific response elements in the DNA (Ahn & Aggarwal Citation2005). The phosphorylation of IκBα is catalyzed by IKK, which is essential for NF-κB activation by most agents. NF-κB has been shown to regulate the expression of a number of genes whose products are involved in tumourigenesis (Aggarwal Citation2004; Ahn & Aggarwal Citation2005). These include anti-apoptotic genes (e.g., ciap, suvivin, traf, cflip, bfl-1, bcl-2 and bcl-xl); cox2; mmp-9; VEGF; genes encoding adhesion molecules, chemokines and inflammatory cytokines; and cell-cycle regulatory genes (e.g., cyclin d1 and cmyc).
We have previously demonstrated that SV, a cholesterol-lowering agent, can potentiate TNF-induced apoptosis through modulation of NF-κB-signalling pathway (Ahn et al. Citation2007) and overcome chemoresistance (Ahn et al. Citation2008a). Additionally, SV suppresses osteoclastogenesis induced by receptor activator of NF-κB ligand (RANKL) and can enhance the anti-cancer effects of capecitabine through suppression of NF-κB-regulated genes (Ahn et al. Citation2008b; Manu et al. Citation2013). Although SV has recently been studied for its potential use in cancer therapy, how combination of SV and BGM can regulates tumour cell apoptosis is poorly established. Because grapefruit juice greatly increases serum concentrations of SV (Lilja et al. Citation1998), we hypothesized that sub-optimal doses of SV and BGM can enhance pro-apoptotic effects by modulating the NF-κB pathway. Our results demonstrate that SV combined with BGM can significantly potentiate TNF-induced apoptosis through down-regulation of NF-κB-regulated antiapoptotic gene products and the NF-κB signalling pathway.
Materials and methods
Chemicals
Bergamottin (BGM) and simvastatin (SV) were purchased from Sigma Aldrich (St. Louis, MO). A solution (50 mM) of BGM and SV was prepared in 100% dimethylsulphoxide, stored at −20 °C and then diluted as needed in cell culture medium. Recombinant human TNF-α was purchased from R&D System (Minneapolis, MN). Penicillin and streptomycin were purchased from Invitrogen (Carlsbad, CA). IMDM medium and FBS were purchased from Lonza (Walkersville, MD). Antibodies against p65, IκBα, cyclin D1, MMP-9, PARP, cIAP-1, Bcl-2, Bcl-xL, Survivin, caspase-3, p21 and p53 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The annexin V staining kit was purchased from BD Biosciences (San Jose, CA). Antibodies to cleaved caspase-3 and p27 were purchased from Cell Signaling Technology (Beverly, MA).
Cell lines
KBM-5 (human chronic myeloid leukaemia) cells were purchased from the American Type Culture Collection. MEF wild-type and MEF p65−/− cells were obtained from the Department of Pharmacology, Yong Loo Lin School of Medicine, National University of Singapore. KBM-5 cells were cultured in IMDM supplemented with 15% FBS. MEF wild-type and MEF p65−/− cells were cultured in DMEM supplemented with 20% FBS.
MTT assay
Cell viability was measured by an MTT assay to detect NADH-dependent dehydrogenase activity. A solution (50 μl) of MTT (5 mg/ml) in 1× phosphate-buffered saline (PBS) was directly added to the cells, which were then incubated for 4 h to allow MTT to metabolize to form formazan. The absorbance was measured with an automated spectrophotometric plate reader at a wavelength of 570 nm. Cell viability was normalized as relative percentages in comparison with untreated controls.
Live/dead cell assay
To assess cytotoxicity, we used a live/dead assay (Invitrogen, Carlsbad, CA), which determines intracellular esterase activity and plasma membrane integrity. In brief, 1 × 106 cells were incubated with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 0.1 nM TNF for 24 h at 37 °C. Cells were stained with the live/dead reagent (5 μM ethidium homodimer and 5 μM calcein-AM) and incubated at 37 °C for 30 min. Cells were analyzed under a fluorescence microscope (FluoView FV1000, Olympus, Tokyo, Japan).
Annexin V assay
To detect apoptosis, we used annexin V antibody conjugated to fluorescein isothiocyanate (FITC). In brief, 1 × 106 cells were incubated with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 0.1 nM TNF for 24 h at 37 °C, and subjected to annexin V staining. Cells were washed, stained with FITC-conjugated antibody to annexin V and analyzed with a flow cytometer (FACS Calibur; BD Biosciences, San Jose, CA).
Cell-cycle analysis
KBM-5 cells were seeded onto 6-well plates at a density of 1 × 106 cells/well and incubated for 2 h. MEF wild-type and MEF p65−/− cells were seeded onto 6-well plates at a density of 1 × 106 cells/well and incubated for overnight. After treatment with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 0.1 nM TNF for 24 h at 37 °C, the cells were collected and washed with 1× PBS. Cell pellets were fixed in 70% cold ethanol overnight at −20 °C. The fixed cells were resuspended in 1 × PBS containing 1 mg/ml RNase A, incubated for 1 h at 37 °C, and the cells were stained by adding 50 μg/ml PI for 30 min at room temperature in the dark. The DNA contents of the stained cells were analyzed using CellQuest pro Software with a FACS Calibur (Becton Dickinson, Heidelberg, Germany) flow cytometry.
Western blot analysis
After cells were treated as indicated with BGM, SV and TNF, the cells were lysed and the total protein concentration was determined by Bradford reagent (Bio-Rad, Hercules, CA). Equal amounts of lysates resolved on sodium dodecyl-polyacrylamide gel electrophoresis (SDS-PAGE) were transferred to a nitrocellulose membrane, and the membrane was blocked with 1× TBS containing 0.1% Tween 20 and 5% skimmed milk or 2% BSA for 1 h at room temperature. After blocking, the membranes were incubated overnight at 4 °C with the respective primary antibodies. The membranes were washed three-times and incubated with diluted horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10 000) for 1 h at room temperature. After three washes, the membranes were detected using the enhanced chemiluminescence (ECL) kit (Millipore, Bedford, MA).
Electrophoretic mobility shift assay (EMSA)
Double-stranded oligonucleotides containing the NF-κB (5′-AGTTGAGGGGACTTTCCCAGGC-3′) or consensus sequences were 5′-end labelled with γ-32P ATP using T4 polynucleotide kinase. Unincorporated nucleotide was removed by passage over a Bio-Gel P-6 spin column (Bio-Rad Inc., Hercules, CA). Nuclear extract was incubated with radiolabelled probe for 20 min, and protein–DNA complexes were separated from free probes by electrophoresis on a 4% native polyacrylamide gel in 0.5 × Tris-HCl (pH 7.5), 1 mM MgCl2, 50 mM NaCl, 0.5 mM EDTA, 4% glycerol, 0.5 mM DTT and 50 mg/ml of poly (dI-dC). The dried gels were visualized, and radioactive bands were quantified by a PhosphorImager, Storm 820 (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK), using ImageQuant software (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK).
Statistical analysis
Statistical analysis was performed by Student’s t-test and one-way analysis of variance (ANOVA). A p value of less than 0.05 was considered statistically significant.
Results
The goal of this study was to investigate the effect of SV combined with BGM on the TNF-signalling pathway that mediates apoptosis and inflammation. Most of the studies were conducted using human chronic myeloid leukaemia KBM-5 cells because these cells express both types of TNF receptors.
SV combined with BGM potentiates apoptosis induced by TNF
Inflammatory cytokines, such as TNF, and chemotherapeutic agents have been shown to activate NF-κB. Because NF-κB activation has been shown to suppress the apoptosis induced by various chemotherapeutic agents (Giri & Aggarwal Citation1998; Wang et al. Citation1998), we examined the effect of SV on apoptosis induced by TNF alone or in combination with BGM. As determined by the MTT assay, SV combined with BGM significantly potentiated the cytotoxic effects of TNF in human chronic myeloid leukaemia cells (KBM-5 and K562) (). To determine whether co-treatment of SV and BGM potentiates apoptosis, we used the live/dead assay, which examines intracellular esterase activity and plasma membrane integrity. SV in combination with BGM enhanced TNF-induced apoptosis in KBM-5 cells (). We next determined the effect of co-treatment of SV and BGM on cell-cycle distribution. We found that the combination therapy induced cell-cycle arrest in the G1/S phase from 42% caused by (SV/TNF) to 55% (BGM/SV/TNF) (). We also used annexin V staining to examine apoptosis by membrane phosphatidylesterase (PS) exposure: from 5% (SV/TNF) to 32% (BGM/SV/TNF) on treatment ().
Figure 1. (A) KBM-5 cells were pretreated with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 1 nM TNF for 24 h. Cell viability was then analyzed by MTT assay. (B) K562 cells were pretreated with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 1 nM TNF for 24 h. Cell viability was then analyzed by the MTT assay. (C) KBM-5 cells (1 × 106 cells/ml) were pretreated with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 1 nM TNF for 24 h. Cell death was determined by the calcein-AM based live/dead assay, as described in “Live/dead cell assay” section. (D) Cells were pretreated with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 1 nM TNF for 24 h. after which the cells were washed, fixed, stained with PI and analyzed for DNA content by flow cytometry. (E) Cells were pretreated with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 1 nM TNF for 24 h. Cells were incubated with FITC-conjugated antibody to annexin V, and then analyzed by flow cytometry for early apoptotic effects.
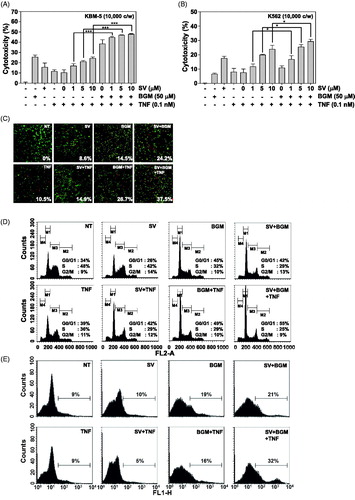
SV combined with BGM suppresses TNF-induced proteins involved in anti-apoptotic and metastatic effect
Because NF-κB regulates the expression of the anti-apoptotic proteins such as cIAP-1, Bcl-2, Bcl-xL and Survivin, we examined whether SV in combination with BGM can suppress the expression of these anti-apoptotic gene products induced by TNF in KBM-5 cells. The results of Western blot analysis showed that TNF induced these anti-apoptotic proteins and that SV in combination with BGM substantially suppressed it (). Expression of cyclin D1, the NF-κB-regulated gene product involved in proliferation, was also suppressed by SV in combination with BGM (). Tumour metastasis is also induced by TNF. MMP-9 has been implicated in invasion and angiogenesis (John & Tuszynski Citation2001). We therefore examined whether SV in combination with BGM can suppress the expression of MMP-9 protein and found that the drug combination can also reduce the expression of MMP-9 (). These results suggest a molecular mechanism that how SV in combination with BGM may possibly potentiate tumour cell apoptosis and metastasis.
Figure 2. (A) KBM-5 cells were treated with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 0.1 nM TNF for 24 h. Whole-cell extracts were prepared, separated on SDS-PAGE and subjected to Western blotting using antibodies against the cIAP-1, Bcl-2, Bcl-xL and Survivin. (B) KBM-5 cells were treated with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 0.1 nM TNF for 24 h. Whole-cell extracts were prepared, separated on SDS-PAGE and subjected to Western blotting using antibodies against the Cyclin D1 and MMP-9. (C) KBM-5 cells were treated with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 0.1 nM TNF for 24 h. Whole-cell extracts were prepared, separated on SDS-PAGE and subjected to Western blotting using antibodies against the p53, p27 and p21. β-Actin was used as a loading control. (D) KBM-5 cells pretreated with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 0.1 nM TNF for 24 h. Whole-cell extract were prepared and analyzed by Western blotting using the caspase-3 antibody. (E) KBM-5 cells were pretreated 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 0.1 nM TNF for 24 h. PARP cleavage was determined by Western blot analysis. β-Actin was used as a loading control. (F) KBM-5 cells pretreated with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 0.1 nM TNF for 30 min. Whole-cell extract were prepared and analyzed by Western blotting using the p-STAT3(Tyr705) and STAT3 antibody.
SV combined with BGM modulates expression of TNF-induced cell-cycle regulatory proteins
p53 protein plays a pivotal role in preventing the onset of cancer and involves the elimination of damaged or infected cells by causing cell-cycle arrest and/or apoptosis (Haupt et al. Citation2003). The CDK inhibitors p21 and p27 play an important role in the regulation of G1/S transition by binding to and inhibiting kinase activity of cyclin D/CDK4 and cyclin E/CDK2, respectively, thus preventing entry of cells into the S phase (Gartel & Tyner Citation2002). To gain further insights into the mechanism(s) of BGM and SV-induced G1/S phase arrest, we determined the effect of this combination on p53, p21 and p27 protein expressions by Western blot analysis and found that the drug combination can substantially increase the expression of p53, p21 and p27 proteins ().
SV combined with BGM potentiates TNF-induced caspase-3 activation and PARP cleavage
TNF binds to TNFR1, which then sequentially recruits TNFR-associated death domain (TRADD) and TNFR-associated factor 2 (TRAF2), leading to the activation of NF-κB and recruitment of Fas-associated death domain, which ultimately leads to the activation of caspases (Wang et al. Citation1998). We investigated whether SV in combination with BGM induced activation of caspase-3. The treatment with SV in combination with TNF and BGM potentiated the activation, as indicated by the presence of cleaved caspase-3 (). We also used the PARP cleavage assay to detect apoptosis in cells treated with the combination of SV and BGM. SV in combination with BGM substantially enhanced PARP cleavage caused by TNF alone as well as SV and TNF combination ().
SV combined with BGM is required for TNF-induced activation of STAT3
We investigated whether SV combined with BGM potentiates TNF-induced constitutive STAT3 activation in KBM-5 cells. The results of Western blot analysis showed that KBM-5 cells exhibited substantially constitutive STAT3 activation and its activation was suppressed by 10 μM SV, but not 50 μM BGM alone. TNF induced stronger STAT3 activation as compared with the basal level of cells and SV in combination with BGM substantially suppressed constitutive STAT3 phosphorylated at the tyrosine 705 site ().
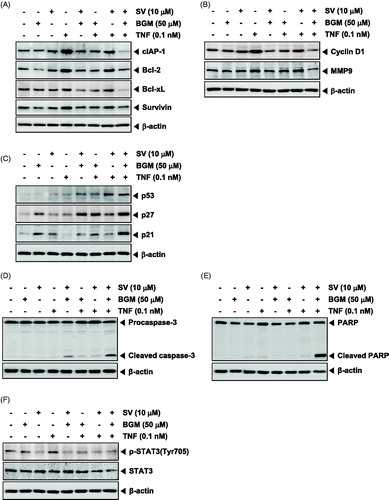
SV in combination with BGM inhibits TNF-induced NF-κB activation
Because the various gene products mentioned above are regulated by NF-κB, we examined whether SV in combination with BGM can also modulate the activation of NF-κB. EMSA revealed that SV and BGM alone had no effect on NF-κB activation. However, SV in combination with BGM exhibited potent inhibitory effects of TNF-mediated NF-κB activation ().
Figure 3. (A) Nuclear extracts were prepared from untreated KBM-5 cells or cells pretreated with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, stimulated 0.1 nM TNF for 30 min, and then assayed for NF-κB binding to DNA by EMSA. (B) Cytoplasmic extracts were prepared from untreated KBM-5 cells or cells pretreated with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 0.1 nM TNF for 30 min, and then subjected to Western blotting using antibody against the IκBα. β-Actin was used as a loading control. (C) Nuclear extracts were prepared from untreated KBM-5 cells or cells pretreated with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, stimulated 0.1 nM TNF for 30 min, and then subjected to Western blotting using antibody against the p65. Lamin B was used as a loading control.
SV in combination with BGM inhibits TNF-dependent IκBα degradation
The translocation of NF-κB to the nucleus is preceded by the phosphorylation, ubiquitination and proteolytic degradation of IκBα (Aggarwal Citation2004). To determine whether inhibition of TNF induced NF-κB activation by SV in combination with BGM is caused by inhibition of IκBα degradation. KBM-5 cells were co-treated with SV and BGM, exposed to TNF, and then examined for IκBα degradation by Western blot analysis using an antibody that recognizes the IκBα. Western blot analysis showed that TNF alone induced IκBα degradation but it could not induce IκBα degradation in combination with both SV and BGM ().
SV in combination with BGM inhibits TNF-induced p65 translocation into the nucleus
We examined whether SV in combination with BGM affects TNF-induced nuclear translocation. Western blot analysis showed that TNF induced nuclear translocation of p65 in KBM-5 cells. When the cells were co-treated with BGM and SV, TNF failed to induce nuclear translocation of p65 ().
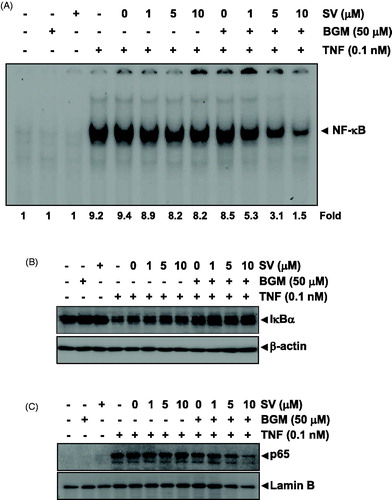
Deletion of p65 abolishes the effect of SV in combination with BGM on TNF-induced apoptosis
The p65 is known to be the active subunit of NF-κB, which regulates anti-apoptotic gene products. As expected, p65 expressed MEF wild-type cells in both cytoplasm and nuclei but not MEF p65−/− cells (). We analyzed MEF wild-type and MEF p65−/− cells to compare the potentiation of TNF-induced apoptosis caused by BGM in combination with SV. We found that BGM in combination with SV potentiated TNF-induced apoptosis in MEF wild type. In MEF p65−/−, lacking functional NF-κB, TNF alone could induce apoptosis. These results indicated that deletion of p65 abolished the potentiation of TNF-induced apoptosis caused by the combination of SV with BGM ().
Figure 4. (A) Cytoplasmic and nuclear extracts were prepared from MEF wild-type and MEF p65−/− cells, separated on SDS-PAGE and electrotransferred to a nitrocellulose membrane. The analysis was performed using p65 antibody. (B) The MEF wild-type and MEF p65−/− (1 × 105 cells/ml) cells were pretreated with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 0.1 nM TNF for 24 h. Cells were stained with live/dead assay reagent for 30 min and then analyzed under a fluorescence microscope. (C) MEF wild-type and MEF p65−/− (1 × 106 cells/ml) cells were treated with 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 0.1 nM TNF for 24 h. Whole-cell extracts were prepared, separated on SDS-PAGE and subjected to Western blotting using antibodies against the Bcl-2 and Survivin. β-Actin was used as a loading control. (F) MEF wild-type and MEF p65−/− (1 × 106 cells/ml) cells were pretreated 10 μM SV in the presence or absence of 50 μM BGM for 12 h, incubated 0.1 nM TNF for 24 h. Caspase-3 cleavage was determined by Western blot analysis. β-actin was used as a loading control.
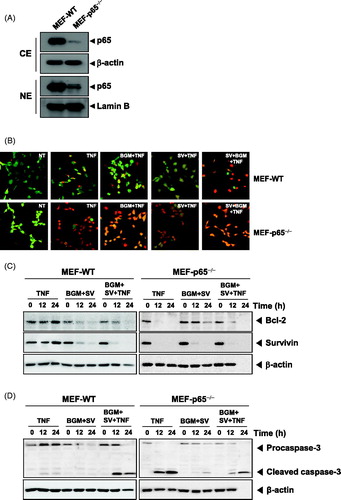
Suppression of TNF-induced expression of anti-apoptotic proteins by SV in combination with BGM requires functional p65
We next determined using both MEF wild-type and MEF p65−/− cells whether the presence of functional p65 is essential for the inhibitory effects of SV in combination with BGM on TNF-induced expression of anti-apoptotic gene products. The results showed that TNF-induced the expression of anti-apoptotic proteins and that SV in combination with BGM inhibited this expression in wild-type MEFs. In MEF p65−/− (, left panels), however, TNF failed to induce expression of anti-apoptotic proteins (, right panels). Also, we investigated whether SV in combination with BGM induces activation of caspase-3 in wild-type MEFs. The treatment with SV in combination with TNF and BGM potentiated the activation, as indicated by the presence of cleaved caspase-3 (, left panels). In MEF p65−/− (, left panels), however, only TNF-treated group induced the activation of caspase-3 due to the loss of p65 function. These results suggested that the presence of functional p65 is essential for the inhibition of anti-apoptotic proteins caused by the drug combination.
Discussion
This study was designed to investigate the potentiated effect of combination therapy of SV and BGM on TNF-induced cell signalling. We found that SV in combination with BGN enhanced cytotoxicity and apoptosis induced by TNF and/or TNF and SV. The combination therapy arrested cells at the S-phase through the downregulation of cyclin D1 and upregulation of p21 and p27 proteins. It inhibited TNF-induced expression of the anti-apoptotic (cIAP-1, Bcl-2, Bcl-xL and Survivin), invasive (MMP-9) and angiogenetic (VEGF) genes. Co-treatment of SV and BGM increased the population of apoptotic cells as demonstrated by annexin V staining and live/dead assays, and induced caspase-3 activation and PARP cleavage. We also found that the combination therapy suppressed TNF-induced NF-κB activation; blocked IκBα degradation; and inhibited p65 translocation into the nucleus as compared with cells treated with TNF and/or TNF and SV. Finally, deletion of p65 gene abolished the potentiation of TNF-induced apoptosis caused by combination of SV with BGM
Our results showed that SV combined with BGM enhanced TNF-induced cytotoxicity and apoptosis of tumour cells by arresting the cells in the S-phase of the cell cycle. Cyclin D1 is required for the progression of cells from the G1 phase to the S-phase of the cell cycle (Matsushime et al. Citation1991), and the combination therapy clearly inhibited its expression in human CML KBM-5 cells. The expression of the cyclin-dependent kinase inhibitors including p21WAF1/CIP1 and p27 which block cell-cycle progression by inhibiting the activity of cyclin/Cdk2 complexes (Nguyen et al. Citation1999; Stewart et al. Citation1999), was also upregulated by SV/BGM/TNF co-treatment.
We found for the first time that the combination therapy (10 μM of SV and 50 μM of BGM) downregulated the expression of anti-apoptotic gene products cIAP-1, Bcl-2, Bcl-xL and Survivin. These observations are in agreement with a report in which SV, 50 μM, was shown to downregulate the expression of Bcl-2 and Bcl-xL but a higher concentration is required to observe maximal inhibitory effect (Ahn et al. Citation2007). Thus, it may be possible that BGM inhibits cytochrome P450 34A enzyme activity which can enhance bioavailability of SV in KBM-5 cells. We also found that SV in combination with BGM inhibited TNF-induced MMP-9 expression, further showing that 50 μM of BGM may have anti-metastatic activity as reported previously in human fibrosarcoma HT-1080 cells (Hwang et al. Citation2010). Moreover, 50 μM of SV has also been reported to inhibit H-Ras-induced invasion, MMP expression and signal transduction in MCF10A breast epithelial cells (Kang et al. Citation2009).
We found for the first time that SV (5–10 μM) in combination with BGM (50 μM) exhibited potent inhibitory effects on TNF-mediated NF-κB activation as compared with the SV alone-treated group. These results are consistent with our previous report in which high concentration of SV (50 μM) was found to suppress TNF-induced NF-κB activation in KBM-5 cells; however, 5 μM SV failed to inhibit TNF-induced NF-κB activation (Ahn et al. Citation2007). It is possible that this particular observation could be attributed to BGM that could enhance the capability of SV to suppress TNF-induced NF-κB activation in our study.
How SV in combination with BGM downregulated the expression of such a wide variety of gene products involved in tumourigenesis, was also investigated. We found for the first time that the employed drug combination is a potent inhibitor of TNF-induced NF-κB activation. Our results are in agreement with prior reports which show that BGM can also suppress PMA-induced NF-κB activation (Hwang et al. Citation2010) and SV can block NF-κB activation induced by various inflammatory stimuli (Ahn et al. Citation2007, Citation2008b; Fraunberger et al. Citation2009). We further observed that TNF-induced IκBα degradation was suppressed by the combination therapy, leading to inhibition of p65 translocation into the nucleus.
Conclusion
Overall, this study showed for the first time that the anti-proliferative and pro-apoptotic effects of the combination therapy consisting of SV and BGM might be mediated through the suppression of NF-κB and NF-κB-regulated gene products. Thus, SV combined with BGM might provide a novel approach to the treatment of CML and potentially other types of cancers that are resistant to chemotherapy as well as radiotherapy.
Funding information
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (NRF-2015R1A4A1042399). This work was also supported by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korean Ministry of Education, Science and Technology (MoEST) (No. 2011-0006220).
Disclosure statement
The authors declare no competing financial interests.
References
- Aggarwal BB. 2003. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 3:745–756.
- Aggarwal BB. 2004. Nuclear factor-kappaB: the enemy within. Cancer Cell. 6:203–208.
- Ahmed T. 2013. High dose simvastatin as a potential anticancer therapy in leukemia patients [theses and dissertations] – Pharmacy, Paper 13. Available from: http://uknowledge.uky.edu/pharmacy_etds/13.
- Ahn KS, Aggarwal BB. 2005. Transcription factor NF-kappaB: a sensor for smoke and stress signals. Ann N Y Acad Sci. 1056:218–233.
- Ahn KS, Sethi G, Aggarwal BB. 2007. Simvastatin potentiates TNF-alpha-induced apoptosis through the down-regulation of NF-kappaB-dependent antiapoptotic gene products: role of IkappaBalpha kinase and TGF-beta-activated kinase-1. J Immunol. 178:2507–2516.
- Ahn KS, Sethi G, Aggarwal BB. 2008a. Reversal of chemoresistance and enhancement of apoptosis by statins through down-regulation of the NF-kappaB pathway. Biochem Pharmacol. 75:907–913.
- Ahn KS, Sethi G, Chaturvedi MM, Aggarwal BB. 2008b. Simvastatin, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, suppresses osteoclastogenesis induced by receptor activator of nuclear factor-kappaB ligand through modulation of NF-kappaB pathway. Int J Cancer. 123:1733–1740.
- Bailey DG, Malcolm J, Arnold O, Spence JD. 1998. Grapefruit juice–drug interactions. Br J Clin Pharmacol. 46:101–110.
- Chan KK, Oza AM, Siu LL. 2003. The statins as anticancer agents. Clin Cancer Res. 9:10–19.
- Fraunberger P, Grone E, Grone HJ, Walli AK. 2009. Simvastatin reduces endotoxin-induced nuclear factor kappaB activation and mortality in guinea pigs despite lowering circulating low-density lipoprotein cholesterol. Shock. 32:159–163.
- Gartel AL, Tyner AL. 2002. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 1:639–649.
- Girennavar B, Jayaprakasha GK, Patil BS. 2007. Potent inhibition of human cytochrome P450 3A4, 2D6, and 2C9 isoenzymes by grapefruit juice and its furocoumarins. J Food Sci. 72:C417–C421.
- Giri DK, Aggarwal BB. 1998. Constitutive activation of NF-kappaB causes resistance to apoptosis in human cutaneous T cell lymphoma HuT-78 cells. Autocrine role of tumor necrosis factor and reactive oxygen intermediates. J Biol Chem. 273:14008–14014.
- Haupt S, Berger M, Goldberg Z, Haupt Y. 2003. Apoptosis-the p53 network. J Cell Sci. 116:4077–4085.
- He K, Iyer KR, Hayes RN, Sinz MW, Woolf TF, Hollenberg PF. 1998. Inactivation of cytochrome P450 3A4 by bergamottin, a component of grapefruit juice. Chem Res Toxicol. 11:252–259.
- Hwang YP, Yun HJ, Choi JH, Kang KW, Jeong HG. 2010. Suppression of phorbol-12-myristate-13-acetate-induced tumor cell invasion by bergamottin via the inhibition of protein kinase Cdelta/p38 mitogen-activated protein kinase and JNK/nuclear factor-kappaB-dependent matrix metalloproteinase-9 expression. Mol Nutr Food Res. 54:977–990.
- John A, Tuszynski G. 2001. The role of matrix metalloproteinases in tumor angiogenesis and tumor metastasis. Pathol Oncol Res. 7:14–23.
- Kang S, Kim ES, Moon A. 2009. Simvastatin and lovastatin inhibit breast cell invasion induced by H-Ras. Oncol Rep. 21:1317–1322.
- Le Goff-Klein N, Koffel JC, Jung L, Ubeaud G. 2003. In vitro inhibition of simvastatin metabolism, a HMG-CoA reductase inhibitor in human and rat liver by bergamottin, a component of grapefruit juice. Eur J Pharm Sci. 18:31–35.
- Lilja JJ, Kivisto KT, Neuvonen PJ. 1998. Grapefruit juice–simvastatin interaction: effect on serum concentrations of simvastatin, simvastatin acid, and HMG-CoA reductase inhibitors. Clin Pharmacol Ther. 64:477–483.
- Manu KA, Shanmugam MK, Li F, Chen L, Siveen KS, Ahn KS, Kumar AP, Sethi G. 2013. Simvastatin sensitizes human gastric cancer xenograft in nude mice to capecitabine by suppressing nuclear factor-kappa B-regulated gene products. J Mol Med (Berl). 92:267–276.
- Matsushime H, Roussel MF, Ashmun RA, Sherr CJ. 1991. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell. 65:701–713.
- Nguyen H, Gitig DM, Koff A. 1999. Cell-free degradation of p27(kip1), a G1 cyclin-dependent kinase inhibitor, is dependent on CDK2 activity and the proteasome. Mol Cell Biol. 19:1190–1201.
- Paine MF, Criss AB, Watkins PB. 2005. Two major grapefruit juice components differ in time to onset of intestinal CYP3A4 inhibition. J Pharmacol Exp Ther. 312:1151–1160.
- Stewart ZA, Leach SD, Pietenpol JA. 1999. p21(Waf1/Cip1) inhibition of cyclin E/Cdk2 activity prevents endoreduplication after mitotic spindle disruption. Mol Cell Biol. 19:205–215.
- Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS, Jr.. 1998. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 281:1680–1683.