
Abstract
Context Shengmai injection (SMI) is a patented Chinese medicine originated from the ancient Chinese herbal compound Shengmai san, which is used extensively for the treatment of cardiovascular and cerebrovascular disease in the clinic.
Objective To determine the neuroprotective effect of SMI, we investigated the effect of SMI on cerebral ischemia/reperfusion (I/R) injury in mice as well as the mechanisms underlying this effect.
Materials and methods Right middle cerebral artery was occluded by inserting a thread through internal carotid artery for 1 h, and then reperfused for 24 h in mice. The neuroprotective effects were determined using transmission electron microscopic examination, the evaluation of infarct volume, neurological deficits and water brain content. Related mechanisms were evaluated by immunofluorescence staining and western blotting. SMI was injected intraperitoneally after 1 h of ischemia at doses of 1.42, 2.84 and 5.68 g/kg. The control group received saline as the SMI vehicle.
Results Results showed that SMI (1.42, 2.84 and 5.68 g/kg) could significantly reduce the infarct volume, SMI (5.68 g/kg) could also significantly improve the neurological deficits, decreased brain water content, as well as the neuronal morphological changes. SMI (5.68g/kg) could significantly inhibit the expression of autophagy-related proteins: Beclin1 and LC3. It also reduced the increase in LC3-positive cells. SMI (5.68 g/kg) remarkably inhibited the phosphorylation of adenosine monophosphate activated protein kinase (AMPK), and down-regulated the phosphorylation of mammalian target of rapamycin (mTOR) and Jun N-terminal kinase (JNK) after 24 h of reperfusion.
Discussion and conclusion The results indicate that SMI provides remarkable protection against cerebral ischemia/reperfusion injury, which may be partly due to the inhibition of autophagy and related signalling pathways.
Introduction
Stroke is the second most common cause of death worldwide, and 80% of stroke cases are ischemic stroke (Macrez et al. Citation2011; Guo et al. Citation2012). Although a number of drugs with potential neuroprotective activities have been used in the treatment of ischemic stroke (Hill et al. Citation2012; Bansal et al. Citation2013), it is still an important cause of mortality and morbidity (Fu et al. Citation2013; Powers et al. Citation2015). It is well established that excitotoxicity, oxidative stress, inflammation and apoptosis are the major pathological mechanisms of ischemia/reperfusion injury (Ozbal et al. Citation2008; Yousuf et al. Citation2009). Many studies have demonstrated the key role of cell death in the neuronal loss and ischemic brain injury (Pamenter et al. Citation2012; Park et al. Citation2012; Winters et al. Citation2013). Apoptotic cell death is more frequently reported, while relatively few cases of autophagic cell death are reported (Rami et al. Citation2008; Winters et al. Citation2013). However, recent studies have shown that autophagy is involved in neural death following cerebral ischemia (Puyal & Clarke Citation2009; Zheng et al. Citation2012).
Autophagy is an intracellular process involving the degradation of macromolecules and organelles (Kost et al. Citation2011). Autophagy may not only help promote cell survival but also promote cell death through excessive self-digestion and degradation of essential cellular constituents (Reggiori & Klionsky Citation2002). In vivo evidence of autophagy has been provided, with a peak at 1d, in a live animal model following cerebral ischemia (Tian et al. Citation2010). Whether autophagy has protective or detrimental effects on brain damage is still unclear. Recent advances have indicated that maintaining a balanced level of autophagy is critically important for neuronal health and function (Chu et al. Citation2009). Autophagy is a tightly regulated pathway including at least five molecular components. This pathway can be stimulated by multiple forms of stress including nutrient or growth factor deprivation, hypoxia, reactive oxygen species (Kroemer et al. Citation2010). Adenosine monophosphate-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) play key roles in mediating autophagy in response to ischemia (Matsui et al. Citation2008). During the reperfusion phase, oxidative stress and endoplasmic reticulum stress occur, followed by the up-regulated expression of Beclin1 and Jun N-terminal kinase (JNK) (De Meyer & Martinet Citation2009). According to the studies described above, modulating autophagy and its related pathways may be beneficial for ischemic stroke.
Shengmai injection (SMI), a modern form of the traditional Chinese formulation Shengmai san, which consists of Radix Ginseng, Radix Ophiopogonis and Fructus Schisandrae, shows significant efficacy for the prevention and treatment of cardiovascular and cerebrovascular diseases in clinic (Wei et al. Citation2011; Chen et al. Citation2012). Many compounds in SMI have been reported to exhibit various potential neuroprotective effects, including the antioxidant effects of Ginsenoside Rg1 (Liu Citation2011), the anti-inflammatory effects of Ginsenoside Re (Lee et al. Citation2012), the anti-apoptotic and anti-autophagic effects of Ginsenoside Rb1 (Chen et al. Citation2010; Lu et al. Citation2011; Hashimoto et al. Citation2012) and the anti-excitotoxic effects of schizandrin (Cheng et al. Citation2008). Previously, we confirmed that SMI inhibited cerebral ischemia/reperfusion-induced brain injury and the expression of tissue factors (Yang et al. Citation2013). However, whether SMI can regulate the autophagy and other related signalling pathways induced by cerebral ischemia/reperfusion has not been demonstrated. Therefore, in the present study, we investigated the effects of SMI against cerebral ischemia/reperfusion-induced autophagy and the AMPK, mTOR and JNK pathways. These findings are expected to provide further evidence for the application of SMI in cerebral diseases such as stroke.
Materials and methods
Chemicals and regents
SMI (Batch No. 10112406) was obtained from SZYY Group Pharmaceutical Ltd. (Jiangyan, Jiangsu, China). 3-Methyladenine (3-MA), and 2,3,5-triphenyltetrazolium chloride (TTC) were purchased from Sigma (St. Louis, MO). The anti-LC3 and anti-Beclin1 antibodies were purchased from Abcam (Cambridge, MA). The antibodies AMPK, p-AMPK, mTOR p-mTOR, JNK and p-JNK antibodies were obtained from Cell Signaling Technology (Danvers, MA). The antibody against GADPH was purchased from KangCheng Bio-tech (Shanghai, China).
Preparation of SMI
SMI was prepared as follows: The powdered Radix Ginseng (100 g) was extracted in a reflux condenser with 90% ethanol at 50 °C 4 times for 2 h each. The extract was combined with a sufficient quantity of activated carbon and adjusted to pH 7. Then water for injection was added for a total volume of 200 mL, and the solution was filtered for dosing. The powdered Fructus Schisandrae (156 g) was first distilled, and 150 mL of the distillate was collected. Then, the dregs of decoction were extracted in a reflux condenser with distilled water at boiling temperature three times for 40 min each. The aqueous extract was filtered and concentrated, then, the extractum was immersed in ethanol two times to ensure the concentration of ethanol was over 85%. The ethanol extract was adjusted to pH 7 and concentrated. Water for injection was added to the extraction for a total volume of 150 mL, and the solution was filtered for dosing. The powdered Radix Ophiopogonis (312 g) was extracted using the same extraction process used for Fructus Schisandrae; 200 mL was obtained and filtered for dosing. Subsequently, the three solution described above were combined with a sufficient quantity of activated carbon and boiled for 30 min. Polysorbate 80 (3–5 g) was added, the solution was filtered, and water for injection was added for a final volume of 1000 mL. Finally, the injection was adjusted to pH 7.5 and sterilized for use (Ministry of Health of the People's Republic of China, 1997).
HPLC fingerprint of SMI
The quality of the SMI used in this study was ensured by HPLC figureprints; therefore, the multiple components of SMI were characterized by an Agilent 1260 HPLC system (Agilent Technologies, Palo Alto, CA) that was equipped with a binary pump, an autosampler, a column oven and a diode-array detector. All chromatographic separations were performed on an Alltima C18 column (4.6 × 250 mm2, 5 μm, Alltech Company, USA) at room temperature. The mobile phase consisted of water (A) and acetonitrile (B). The following gradient program was used: 0–10 min, 5–15% B; 10–30–min, 15–35% B; 30–50–min, 35–43% B; and 50–80–min, 43–65% B. The flow rate was 1–mL/min, and the UV detection wavelength was set at 203–nm. The column temperature was 25–°C, and the sample injection volume was 20–μL. The chromatographic data were recorded and processed with an Agilent Chemstation B.04.03.
Animals and drug administration
Male C57BL/6J mice weighing 18–22 g were provided by the Model Animal Research Centre of Yangzhou University (Yangzhou, Jiangsu, P.R. China). The mice were housed under controlled environmental conditions, including ambient temperature (20 ± 2 °C), a relative humidity of 55–65%, and a 12 h light/dark cycle. The C57BL/6J mice were randomly divided into several groups, including sham, model and drug treatments groups (n = 6). SMI, at three doses (1.42, 2.84 and 5.68 g crude drug/kg (which are converted based on the clinical dosages), and 3-methyladenine (3-MA, Sigma, M9281), at 300 μg/kg (Lu et al. Citation2011) were injected intraperitoneally after 1 h of ischemia. The other groups were treated with vehicle (0.9% NaCl). All of the experiments and animal care were performed in accordance with the Provisions and General Recommendations of the Chinese Experimental Animal Administration Legislation and the related ethical regulations on animal research (National Research Council of US, 1996).
Transient middle cerebral artery occlusion
All mice were maintained under anaesthesia with chloral hydrate (400 mg/kg), which was administered intraperitoneally. MCAO was performed according to a previously described method (Khayyam et al. Citation1999). A blunt-tip 6-0 nylon suture with silicone coating on its head was inserted into the internal carotid artery (ICA) through the external carotid artery (ECA) stump and gently advanced to the Circle of Willis to occlude the right middle cerebral artery (MCA). The suture was withdrawn after an ischemic period of 1 h to induce reperfusion. During the surgical procedure, the rectal temperature was maintained at 37.0 ± 0.5 °C using a heating plate. Mice in the sham group underwent the same surgical procedures except for the MCAO.
Evaluation of infarct volume, neurological deficits and brain water content
After 24 h of reperfusion, the neurological deficits of the experimental animals (6 animals/group) were graded on an 18-point scale as previously described (Garcia et al. Citation1995). After scoring, the mice were executed and the brains were rapidly removed and frozen for 10 min at −20 °C to evaluate the infarct volume. The brains were cut into 2 mm coronal sections, and the sections were immersed in 1% TTC for 30 min at 37 °C. The infarct area was measured using NIH Image J software (Version 1.42, National Institutes of Health, MD, USA). The infarct volume was calculated by taking the average of the infarct area on both sides of the slice and multiplying it by the section thickness. Brain water contents (6 animals/group) were determined 24 h after reperfusion. Infarct brain hemispheres were quantified with electronic scale (wet weight) and dried overnight at 105 °C in a desiccating oven and then weighted (dry weight). The total brain water was calculated according to [(wet weight − dry weight)/wet weight] × 100% as described previously (Jiang et al. Citation2014).
Transmission electron microscopic examination
Mice (3 animals/group) were deeply anesthetized and perfused with 50 mL of normal saline, followed by 0.1 M phosphate buffer (PBS, pH = 7.4) that contained 4% paraformaldehyde and 0.25% glutaraldehyde. The brains were removed and kept in 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M phosphate buffer (PBS, pH = 7.4) overnight. A small amount of brain tissue was taken from the ischemic penumbra of the cortex the next day and cut into small pieces of 1 mm × 1 mm × 1 mm. The pieces were rinsed three times with 0.1 mol/L PBS and immersed in 1% osmium tetroxide in 0.1 mol/L PBS for 2 h at 4 °C. After that, the tissue block was dehydrated in graded ethanol solutions and embedded in epoxy resin. Polymerization was performed at 80 °C for 24 h. Blocks were cut on a Reichert ultramicrotome into ultrathin sections (60–70 nm), stained with 3% lead citrate, and subsequently examined using a transmission electron microscope (JEOL, JEM-2000EX).
Immunofluorescence staining
Anesthetized mice (3 animals/group) were perfused with 50 mL normal saline, followed by 0.1 M phosphate buffer (pH 7.4) containing 4% paraformaldehyde. The brains were removed, postfixed overnight in 4% paraformaldehyde and immersed in a 30% sucrose solution until they were observed to sink in the solution. Then, the brains were cut into 20 μm coronal sections and blocked with 3% normal donkey serum that contained 0.3% Triton X-100 for 1 h at room temperature. They were then incubated at 4 °C with an LC3 primary antibody (1:200, Abcam, ab64781) or an Beclin1 primary antibody (1:200, Abcam, ab55878) for 48 h. Washed three times with phosphate buffer, they were incubated with secondary antibody (Alexa Fluor 488, 1:800, Invitrogen, A21206) for 3 h at room temperature. Last, the sections were coverslipped using an anti-fading agent (Beyotime, P0126, Shanghai, China). Fluorescence signals were detected using the confocal laser scanning microscopy (Olympus, FV1000, Tokyo, Japan). The LC3 and Beclin1 positive cells were counted in 10 non-overlapping high power fields covering the majority of cortex each section.
Western blotting
After ischemia/reperfusion, brain tissue (4 animals/group) from the ischemic cortex within the territory of the right MCA and from the corresponding area of the sham-operated mice was homogenized, and proteins were extracted with a lysis buffer (Beyotime, P0013) containing 1% phenylmethylsulfonyl fluoride and 1% Halt Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific Inc,78420). Then, 60μg protein aliquots from each sample were separated via SDS-PAGE, and transferred to Immobilon-P transfer membranes. The membranes were blocked in 3% bovine serum albumin (BSA) in Tris-buffered saline with 1% Tween buffer for 1.5 h and then incubated overnight at 4 °C with the following primary antibodies: anti-LC3 (1:1000), anti-Beclin1 (1:500), anti-mTOR (1:1000), anti-phospho-mTOR (1:500), anti-AMPKα (1:1000), anti-phospho-AMPKα (1:500) anti-JNK1 (1:1000), anti-phospho-JNK (1:500) and anti-GAPDH (1:5000). Then, the membranes were washed and incubated with horseradish peroxidase-conjugated secondary antibodies (1:10,000, Bioworld, St. Louis Park, MN, anti-rabbit, BS10350, anti-mouse, BS12478) for 1.5 h at room temperature. After the membranes were washed three times with TBST, the protein bands were detected with ECL detection reagent (Beyotime, P0018, Shanghai, China), visualized using a UVP BIOSPECTRUM (Upland, CA) imaging system and quantified by Quantity One software 4.6.2. GAPDH was used as the loading control according to the previous reports (Cui et al. Citation2012). The level of each sample was first normalized against the GAPDH level of the corresponding sample. Then, we normalized it against the level of a sham sample according to a previous report (Huang et al. Citation2012).
Statistical analysis
The results were expressed as the mean ± S.D. The data were analyzed by Student’s two-tailed-t-test for comparisons between two groups and by one-way ANOVA, followed by Dunnett’s test when the data involved three or more groups. A value of p< 0.05 was considered statistically significant (Gu et al. Citation2012).
Results
HPLC analysis of Shengmai injection
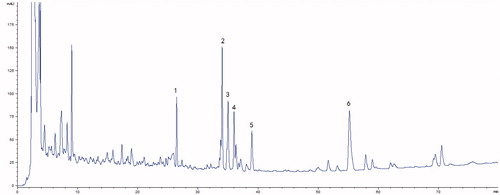
The typical HPLC fingerprint of SMI is shown in . By comparing both the retention times and the UV spectra of the reference standards, 6 compounds [(1) Ginsenoside Re, (2) Ginsenoside Rb1, (3) Ginsenoside Rc, (4) Ginsenoside Rb2, (5) Ginsenoside Rd and (6) Schizandrol A] were definitively identified in SMI.
Figure 1. HPLC fingerprint chromatogram of Shengmai injection at 203 nm. (1) Ginsenoside Re, (2) Ginsenoside Rb1, (3) Ginsenoside Rc, (4) Ginsenoside Rb2, (5) Ginsenoside Rd and (6) Schizandrol A.
Effects of SMI on infarct volume, neurological deficit score and brain water content following ischemia/reperfusion injury
shows photographs of typical coronal sections from each group. Compared with the sham group (4.66 ± 0.64%), the infarct volume of the model group was significantly increased (40.81 ± 4.68%, p< 0.01). Treatment with SMI at all doses significantly reduced the infarct volume in the SMI-treated groups compared with the model group. The protective effect of SMI at 5.68g/kg was the most potent of all the treatments. In addition, 3-MA, a type of autophagy inhibitor that was used as a positive control, also significantly reduced the infarct volume (p< 0.01, ).
Figure 2. Effects of Shengmai injection (SMI) on the infarct volume (A, B), the neurological scores (C) and the brain water content in C57 mice subjected to tMCAO/R. Mice were subjected to 1 h of ischemia and 24 h of reperfusion (tMCAO/R). SMI (ip) was administered after 1h ischemia. 3-Methyladenine (ip) was administered at the onset of the surgery as a positive control. Images of TTC-stained brain sections (A) from Vehicle- or SMI-treated mice 24h after reperfusion. Infarct volume (B), neurological scores (C) and brain water content (D) were evaluated for Vehicle- or SMI-treated mice 24 h after reperfusion. The data are the mean ± SD, n = 6. ##p < 0.01 versus sham-operated mice, **p < 0.01 versus tMCAO/R mice.
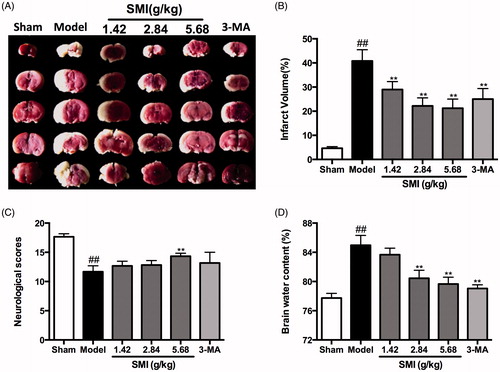
The neurological scores, which were evaluated at 24 h after reperfusion in the sham, model and drug-treated groups (n = 6 in each group), are shown in . The scores in the group treated with 5.68 g/kg (14.4 ± 0.52) were significantly better than those in the model group (11.60 ± 1.03, p< 0.01). The other SMI-treated group and the 3-MA treated group also exhibited improved neurological deficits compared to those in the tMCAO/R mice, but without statistical difference.
Brain water content was quantitatively determined to evaluate the effects of SMI following focal ischemia/reperfusion in the sham, model, and drug-treated groups (n = 6 in each group), are shown in . Results demonstrated that compared with the sham group, brain water content was significantly increased in the model group (84.9 ± 0.55%, p< 0.01). Treatment with SMI at the dose of 2.84 g/kg (80.4 ± 0.44%, p< 0.01) and 5.68 g/kg (79.6 ± 0.38%, p < 0.01) could significantly reduce the brain water content in these two SMI-treated groups compared with the model group. In addition, the group treated with 3-MA could also significantly reduced the infarct volume (79.1 ± 0.20, p< 0.01).
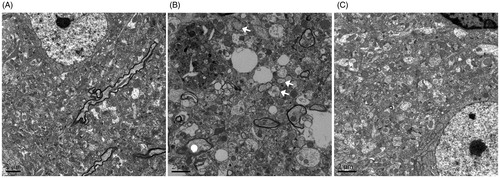
Effects of SMI on the on the formation of autophagosomes following ischemia/reperfusion injury
shows the morphological changes of cortical neurons, as observed under the electron microscope. The neurons in the sham group appeared normal, with relatively healthy-looking endoplasmic reticulum, mitochondria, lysosomes and the nuclei. After 1 h of ischemia and 24h of reperfusion, the neurons were moderately vacuolated, the mitochondria were broken, and many lysosomes and autophagosomes could be found in neurons. However, when the mice were treated with SMI, the neuronal injury was alleviated, and the numbers of lysosomes and autophagosomes were decreased; this effect was also observed in the 3-MA treatment group.
Figure 3. Effects of Shengmai injection (SMI) on the neuron’s morphological changes. Representative electron microphotographs showing neuronal injury in the ischemic penumbra of the cerebral cortex. (A) Mice subjected to the sham operation. (B) Mice subjected to 1h of ischemia and 24 h of reperfusion (tMCAO/R). (C) Mice subjected to 1 h of ischemia and 24h of reperfusion (tMCAO/R). SMI (5.68 g/kg, ip) was administered after 1h of ischemia. Scale bars = 2 μm.
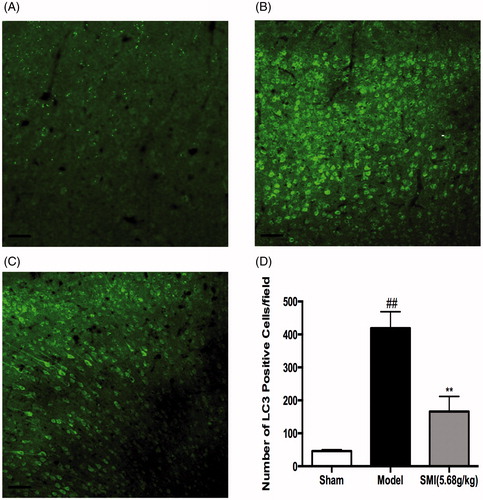
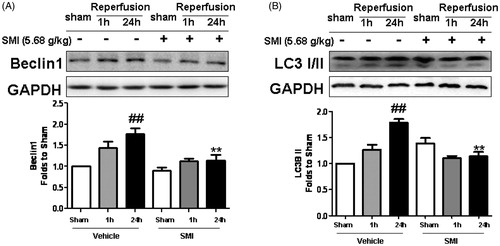
Effects of SMI on the protein levels of LC3 and Beclin following ischemia/reperfusion injury
LC3 and Beclin1 are biomarkers for autophagy activation in mammalian cells. To examine autophagic activity after tMCAO/R and to determine whether SMI could inhibit the activation of autophagy, we performed immunofluorescence staining for LC3 in the cortex in the mice of the sham, model and SMI-treated groups. Confocal laser scanning photomicrographs showed that the LC3-positive cells were mainly present in the ischemic penumbra of the cortex after 1h ischemia/24 h reperfusion; this area was close to the core of ischemia, as reported previously (Xu & Zhang Citation2011). Compared with the number in the sham group, the number of LC3-positive cells was significantly increased in the model group. Following SMI treatment, the number of LC3-positive cells in the high-dose treatment group was markedly decreased compared to that in the model group ().
Figure 4. Effects of Shengmai injection (SMI) on the expression of LC3 in brain sections. Representative microphotographs showing immunofluorescence staining for LC3. (A) Mice subjected to the sham operation. (B) Mice subjected to 1h of ischemia and 24h of reperfusion (tMCAO/R). (C) Mice subjected to 1h of ischemia and 24 h of reperfusion (tMCAO/R). SMI (5.68 g/kg, ip) was administered after 1 h of ischemia. Representative brain sections from the ischemic penumbra of the cortex are shown. Scale bars = 50 μm. (D) Quantification of LC3 positive cells in the ischemic penumbra of the cortex (10f fields/brain). n = 3 per group. ##p < 0.01 versus sham-operated mice, **p < 0.01 versus tMCAO/R mice.
In addition, we also tested the protein expression of LC3 and Beclin1 in the ischemic cortex. We found that the level of Beclin1 following 1h ischemia/24 h reperfusion was significantly greater than that following the sham treatment and that SMI could significantly inhibit the increased expression of Beclin1 following 1 h ischemia/24 h reperfusion (p < 0.01, ). Western blot analysis revealed that the LC3-II was significantly up-regulated in the tMCAO/R mice while SMI significantly attenuated the 1h ischemia/24 h reperfusion-elicited LC3-II up-regulation (p < 0.01, ).
Figure 5. Effects of Shengmai injection (SMI) on the expression of Beclin1 (A) and LC3 (B) in C57 mice subjected to tMCAO/R. The mice were subjected to 1h of ischemia and 24 h of reperfusion (tMCAO/R). SMI (5.68g/kg, ip) was administered after 1 h of ischemia. Representative Western blot analyses showing the levels of Beclin1 and LC3. GAPDH is shown to verify protein loading. The data are the mean ± SD, n = 3. ##p < 0.01 versus sham-operated mice, **p < 0.01 versus tMCAO/R mice.
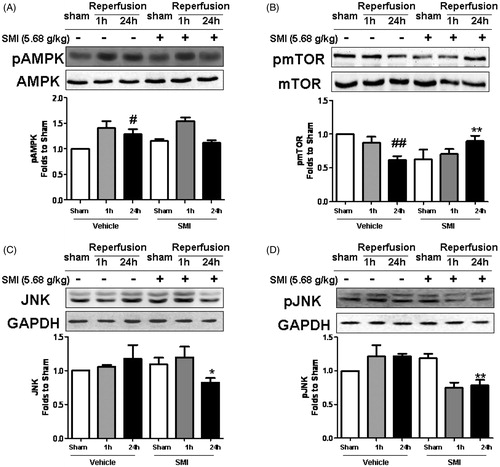
Effects of SMI on AMPK-mTOR and JNK pathways following ischemia/reperfusion injury
To investigate the possible mechanism of SMI-mediated inhibition of autophagy, we further evaluated its effects on the AMPK-mTOR pathway. show that compared with the levels in the sham group, the level of phosphorylated AMPKα was markedly increased (p< 0.05), while the level of phosphorylated mTOR was significantly decreased (p< 0.01) following 1 h ischemia/24 h reperfusion. After SMI treatment, both of them tended to return to normal: the level of phosphorylated AMPKα was decreased and phosphorylated mTOR was increased (p< 0.01). Moreover, we also demonstrated the effect of SMI on JNK and its phosphorylation. Western blot analysis showed that both JNK and p-JNK were increased following 1 h ischemia/24 h reperfusion, and that SMI could significantly attenuate their up-regulation (p< 0.05 and p < 0.01, ).
Figure 6. Effects of Shengmai injection (SMI) on the expression and phosphorylation of AMPK, mTOR and JNK in C57 mice subjected to tMCAO/R. The mice were subjected to 1h of ischemia and 24 h of reperfusion (tMCAO/R). SMI (5.68g/kg, ip) was administered after the 1 h of ischemia. Representative Western blot analyses showing phosphorylated and total levels of AMPK, mTOR, JNK. GAPDH is shown to verify protein loading. The data are the mean ± SD, n = 3. #p < 0.05, ##p < 0.01 versus sham-operated mice, *p < 0.05, **p < 0.01 versus tMCAO/R mice.
Discussion
SMI is a modern form of the traditional Chinese formula Sheng-Mai-San. It has long been used to treat the loss of essence-energy and excessive body fluid and is especially prescribed for cardiovascular and cerebrovascular diseases in China (Wang et al. Citation2002; Lee et al. Citation2005). The present study confirmed the neuroprotective potential of SMI at doses of 1.42, 2.84 and 5.68 g/kg (crude drug). SMI could remarkedly ameliorated the abnormalities in infarct volume, neurological scores and brain water content after 1 h of ischemia followed by 24 h of reperfusion; these effects were similar to those of 3-MA (a type of autophagy inhibitor). We found that SMI could inhibit autophagy and modulate focal cerebral ischemia/reperfusion injury-induced effects on related pathways.
Ischemic stroke develops from a complex cascade of pathophysiological events. Processes of excitotoxicity, oxidative stress, inflammation and apoptosis have been proposed to occur within the peri-infarct area (Zhang et al. Citation2004; Wong & Crack Citation2008; Szydlowska & Tymianski Citation2010). Recent studies have shown that autophagy is involved in the neural death pathway that follows cerebral ischemia, and scientists have already monitored autophagic processes in vivo in live stroke patients (Tian et al. Citation2010). The role of autophagy in ischemic stroke is still debated: it has both protective and detrimental effects (Sheng et al. Citation2010; Yan et al. Citation2011; Gao et al. Citation2012). It has been reported that Ginsenoside Rb1, one of major compounds in SMI, could alleviate ischemia/reperfusion insults in rats and that an autophagic mechanism was involved in the benefits (Yang et al. Citation2013); these findings indicated that SMI might have a similar mechanism against ischemic stroke.
The cellular events that occur during autophagy follow distinct stages: autophagy induction, vesicle nucleation, vesicle expansion and completion, lysosome fusion and breakdown of its contents inside the autolysosome (Levine & Kroemer Citation2008). In our study, a remarkable increase in lysosome number was observed in the model group compared to the sham-operated group. Administration of SMI after ischemia could inhibit the excessive formation of lysosomes. On the other hand, we also administrated 3-MA (an autophagy inhibitor) as a positive control, and confirmed that 3-MA could also improve ischemia/reperfusion injury. Our findings suggest that SMI ameliorated focal cerebral ischemia/reperfusion injury by inhibiting autophagy.
Beclin1 and LC3 are crucial proteins for autophagy. Beclin1 (Atg6) is a well-known key regulator of autophagy. It governs most autophagy processes and has been implicated in endocytic trafficking, phagocytosis, cytokinesis control and pollen germination (Wirawan et al. Citation2012). Regarding LC3, a C-terminal fragment of LC3 is cleaved immediately after LC3 is synthesized, yielding a cytosolic form called LC3-I (18 kD). A subest of LC3-I is further converted to an autophagosome associating form, LC3-II (16 kD). LC3-II is stably associated with the autophagosome membrane, and its biochemical and microscopic detection is widely used to measure cellular autophagy (Mizushima et al. Citation2010). In our experiment, both Beclin1 and LC3 were significantly increased in the ischemia/reperfusion group, and SMI treatment could decrease the expression of Beclin1 and LC3 as well as the level of LC3 binding to the autophagic membranes. These results could further indicate that the neuroprotective effect of SMI was related to autophagy.
Autophagy is an evolutionarily conserved pathway involving lysosomal degradation of cytoplasmic organelles or cytosolic components. This process occurs at a basal level and is regulated by numerous different signalling pathways (Yang & Klionsky Citation2010). The following two complexes are well known to regulate autophagy: the Atg1/unc-51-like kinase (ULK) complex and the Beclin1/class III phosphatidylinositol 3-kinase (PI3K) complex. The ULK complex plays a critical role in autophagy induction, while Beclin1 interacts with several enhancing or inhibitory factors that modulate its binding to Vps34, the catalytic unit of PI3K, whose lipid kinase activity is essential for autophagy (Kroemer et al. Citation2010). AMPK and mTOR have been reported to regulate autophagy through direct phosphorylation of ULK1 (Kim et al. Citation2011). In stroke models, AMPK is related to the regulation of hypothalamic energy balance (McCullough et al. Citation2005; Li & McCullough Citation2010), while mTOR plays a very important role in protein synthesis and apoptosis (Koh et al. Citation2008; Koh Citation2008). Meanwhile, JNK promotes the phosphorylation of C-Jun and further increases the mRNA expression of Beclin1 (Kiyono et al. Citation2009). JNK1-mediated multi-site phosphorylation of Bcl-2 stimulates starvation-induced autophagy by disrupting the Bcl-2/Beclin1 complex (Wei et al. Citation2008). It is also reported that JNK inhibitors could significantly suppress the expression of Beclin1 in a stroke model (Liu et al. Citation2012). According to the previous reports, the induction of the phosphorylation of mTOR at Ser 2448 could continue to increase at 24 h after reperfusion and could be detected at 48 or 72 h after reperfusion (Ayuso et al. Citation2010; Xie et al. Citation2013). The phosphorylation of AMPK was transiently increased at 24 h after ischemia/reperfusion (Nam et al. Citation2013).
In addition, the expression of JNK and its activation were also significantly increased at 24h reperfusion after cerebral ischemia (Huang et al. Citation2013). Thus, we examined three complexes related pathways at 24 h after reperfusion to investigate the effect of SMXZF on cerebral ischemia/reperfusion injury-induced autophagy. To further investigate the effect of SMI on ischemia/reperfusion-induced autophagy, we examined three complexes-related pathways. Our research showed that compared with the levels in the vehicle-treated group, the phosphorylation of AMPKα at Thr 172 was decreased in the SMI-treated groups, while the phosphorylation of mTOR at Ser 2448 was increased. In this study, we also tested the protein level of JNK and its phosphorylation and found that SMI could inhibit both the expression of JNK and its phosphorylation following 1 h ischemia/24h reperfusion in mice. Hence, we demonstrated that SMI-suppressed autophagy was linked with modulation of the AMPK-mTOR and JNK pathways.
It should be noted that there were some limitations to this study. First, further studies are needed to investigate the other signalling pathways by which SMI may regulate ischemia/reperfusion-induced autophagy. Second, excitotoxicity, oxidative stress, inflammation and apoptosis are also involved in cerebral ischemia/reperfusion injury. Whether these mechanisms are responsible for the neuroprotective effects of SMI still needs to be further explored. Former studies have shown that some components from SMI such as Ginsenoside Rb1, Re may inhibit neuronal apoptosis and related signalling pathways (Xu et al. Citation2005; Yuan et al. Citation2007), and recent study from our lab have shown that a cocktail of four active components derived from Sheng Mai San, named SMXZF (Guo et al. Citation2014), could inhibited hydrogen peroxide-induced PC12 cell apoptosis by inhibiting the caspase-3/ROCK1/MLC pathway (Shen et al. Citation2015), so it is probably that SMI may also exerted its neuroprotective effects by inhibit apoptosis and related signalling pathways, which need further confirmation in the future. In addition, the best time window for SMI treatment must also be studied in the future.
It should also be noted that the major components contained in SMI are saponins with relatively large molecular weight and amphiphilic molecular structure, which are usually difficult to penetrate the blood–brain barrier. However, studies have demonstrated that the distribution of Ginsenoside Rb1, Rg1, Re, Rd and Notoginsenoside R1 in cerebral ischemia brain was much higher than that of normal ones (Wang et al. Citation2015). And whether the SMI may have direct effects on blood–brain barrier still needs to be studied further.
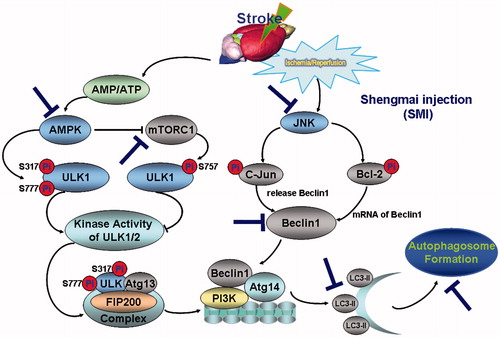
In conclusion, in this study, we demonstrate that SMI is effective for the treatment of cerebral ischemia/reperfusion injury in mice, which and that this effect is linked with the inhibition of autophagy and modulation of the AMPK-mTOR and JNK pathways (a schematic diagram of the effects of SMI against cerebral ischemia/reperfusion injury is shown in ). Our findings provide some new insight into its molecular mechanisms for the treatment of cerebrovascular diseases and a pharmacological basis for expanding the clinical applications of SMI.
Figure 7. Schematic diagram of the protective effect of Shengmai injection (SMI) against cerebral ischemia/reperfusion in mice via autophagy and related signalling pathways. SMI could remarkably inhibit the phosphorylation of AMPK and down-regulated the phosphorylation of mTOR and JNK after 24 h of reperfusion.SMI could significantly inhibit the autophagy-related proteins including Beclin 1 and LC3, and then decrease the formation of autophagosome so as to suppress the cerebral ischemia/reperfusion in mice.
Funding information
This research was supported by the National Natural Science Foundation of China (No. 81274004&81274131), National Key Technologies R&D Program of China (No. 2008BAI51B03), 2011 Program for Excellent Scientific and Technological Innovation Team of Jiangsu Higher Education, and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Acknowledgements
The authors are grateful to Dr. Wang Wen, Department of Human Anatomy and Embryology, The Fourth Military Medical University, China, for his technical assistance for the LC3 immunofluorescence experiments.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Ayuso MI, Hernández-Jiménez M, Martín ME, Salinas M, AlcÃzar A. 2010. New hierarchical phosphorylation pathway of the translational repressor eIF4E-binding protein 1 (4E-BP1) in ischemia-reperfusion stress. J Biol Chem. 285:34355–34363.
- Bansal S, Sangha KS, Khatri P. 2013. Drug treatment of acute ischemic stroke. Am J Cardiovasc Drugs. 13:57–69.
- Chen J, Yao Y, Chen H, Kwong JS, Chen J. 2012. Shengmai (a traditional Chinese herbal medicine) for heart failure. Cochrane Database Syst Rev. 11:CD005052
- Chen Z, Lu T, Yue X, Wei N, Jiang Y, Chen M, Ni G, Liu X, Xu G. 2010. Neuroprotective effect of ginsenoside Rb1 on glutamate-induced neurotoxicity: with emphasis on autophagy. Neurosci Lett. 482:264–268.
- Cheng HY, Hsieh MT, Wu CR, Tsai FH, Lu TC, Hsieh CC, Li WC, Lin YT, Peng WH. 2008. Schizandrin protects primary cultures of rat cortical cells from glutamate-induced excitotoxicity. J Pharmacol Sci. 107:21–31.
- Chu CT, Plowey ED, Dagda RK, Hickey RW, Cherra SJ 3rd, Clark RS. 2009. Autophagy in neurite injury and neurodegeneration: in vitro and in vivo models. Meth Enzymol. 453:217–249.
- Cui D, Wang L, Qi A, Zhou Q, Zhang X, Jiang W. 2012. Propofol prevents autophagic cell death following oxygen and glucose deprivation in PC12 cells and cerebral ischemia-reperfusion injury in rats. PLoS One. 7:e35324.
- De Meyer GR, Martinet W. 2009. Autophagy in the cardiovascular system. Biochim Biophys Acta. 1793:1485–1495.
- Fu P, Peng C, Ding JY, Asmaro K, Sullivan JM, Guthikonda M, Ding Y. 2013. Acute administration of ethanol reduces apoptosis following ischemic stroke in rats. Neurosci Res. 76:93–97.
- Gao L, Jiang T, Guo J, Liu Y, Cui G, Gu L, Su L, Zhang Y. 2012. Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PLoS One. 7:e46092.
- Garcia JH, Wagner S, Liu KF, Hu XJ. 1995. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats Statistical validation. Stroke. 26:627–634.
- Gu JH, Ge JB, Li M, Wu F, Zhang W, Qin ZH. 2012. Inhibition of NF-κB activation is associated with anti-inflammatory and anti-apoptotic effects of Ginkgolide B in a mouse model of cerebral ischemia/reperfusion injury. Eur J Pharm Sci. 47:652–660.
- Guo C, Tong L, Xi M, Yang H, Dong H, Wen A. 2012. Neuroprotective effect of calycosin on cerebral ischemia and reperfusion injury in rats. J Ethnopharmacol. 144:768–774.
- Guo Z, Cao G, Yang H, Zhou H, Li L, Cao Z, Yu B, Kou J. 2014. A combination of four active compounds alleviates cerebral ischemia reperfusion injury in correlation with inhibition of autophagy and modulation of AMPK/mTOR and jnk pathways. J Neurosci Res. 92:1295–1306.
- Hashimoto R, Yu J, Koizumi H, Ouchi Y, Okabe T. 2012. Ginsenoside Rb1 prevents MPP(+)-induced apoptosis in PC12 cells by stimulating estrogen receptors with consequent activation of ERK1/2, Akt and inhibition of SAPK/JNK, p38 MAPK. Evid Based Complement Alternat Med. 2012:693717.
- Hill MD, Martin RH, Mikulis D, Wong JH, Silver FL, Terbrugge KG, Milot G, Clark WM, Macdonald RL, Kelly ME, et al. 2012. Safety and efficacy of NA-1 in patients with iatrogenic stroke after endovascular aneurysm repair (ENACT): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 11:942–950.
- Huang J, Li Y, Tang Y, Tang G, Yang GY, Wang Y. 2013. CXCR4 antagonist AMD3100 protects blood-brain barrier integrity and reduces inflammatory response after focal ischemia in mice. Stroke. 44:190–197.
- Huang P, Zhou CM. Qin-Hu, Liu YY, Hu BH, Chang X, Zhao XR, Xu XS, Li Q, Wei XH. 2012. Cerebralcare Granule(R) attenuates blood-brain barrier disruption after middle cerebral artery occlusion in rats. Exp Neurol. 237:453–463.
- Jiang M, Li J, Peng Q, Liu Y, Liu W, Luo C, Peng J, Li J, Yung KK, Mo Z. 2014. Neuroprotective effects of bilobalide on cerebral ischemia and reperfusion injury are associated with inhibition of pro-inflammatory mediator production and down-regulation of JNK1/2 and p38 MAPK activation. J Neuroinflammation. 11:167
- Khayyam N, Thavendiranathan P, Carmichael FJ, Kus B, Jay V, Burnham WM. 1999. Neuroprotective effects of acetylsalicylic acid in an animal model of focal brain ischemia. Neuroreport. 10:371–374.
- Kim J, Kundu M, Viollet B, Guan KL. 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 13:132–141.
- Kiyono K, Suzuki HI, Matsuyama H, Morishita Y, Komuro A, Kano MR, Sugimoto K, Miyazono K. 2009. Autophagy is activated by TGF-beta and potentiates TGF-beta-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. 69:8844–8852.
- Koh PO. 2008. Melatonin prevents ischemic brain injury through activation of the mTOR/p70S6 kinase signaling pathway. Neurosci Lett. 444:74–78.
- Koh PO, Cho JH, Won CK, Lee HJ, Sung JH, Kim MO. 2008. Estradiol attenuates the focal cerebral ischemic injury through mTOR/p70S6 kinase signaling pathway. Neurosci Lett. 436:62–66.
- Kost A, Kasprowska D, Labuzek K, Wiaderkiewicz R, Gabryel B. (2011). Autophagy in brain ischemia. Postȩpy Hig Med Dośw [Online].Avaliable at:http://www.researchgate.net/publication/51630233.Accessed on 13 August 2015.
- Kroemer G, Mariño G, Levine B. 2010. Autophagy and the integrated stress response. Mol Cell. 40:280–293.
- Lee IY, Lee CC, Chang CK, Chien CH, Lin MT. 2005. Sheng mai san, a Chinese herbal medicine, protects against renal ischaemic injury during heat stroke in the rat. Clin Exp Pharmacol Physiol. 32:742–748.
- Lee KW, Jung SY, Choi SM, Yang EJ. 2012. Effects of ginsenoside Re on LPS-induced inflammatory mediators in BV2 microglial cells. BMC Complement Altern Med. 12:196.
- Levine B, Kroemer G. 2008. Autophagy in the pathogenesis of disease. Cell. 132:27–42.
- Li J, McCullough LD. 2010. Effects of AMP-activated protein kinase in cerebral ischemia. J Cereb Blood Flow Metab. 30:480–492.
- Liu L, Fang YQ, Xue ZF, He YP, Fang RM, Li L. 2012. β-Asarone attenuates ischemia-reperfusion-induced autophagy in rat brains via modulating JNK, p-JNK, Bcl-2 and Beclin 1. Eur J Pharmacol. 680:34–40.
- Liu Q, Kou JP, Yu BY. 2011. Ginsenoside Rg1 protects against hydrogen peroxide-induced cell death in PC12 cells via inhibiting NF-κB activation. - activation. Neurochem Int. 58:119–125.
- Lu T, Jiang Y, Zhou Z, Yue X, Wei N, Chen Z, Ma M, Xu G, Liu X. 2011. Intranasal ginsenoside Rb1 targets the brain and ameliorates cerebral ischemia/reperfusion injury in rats. Biol Pharm Bull. 34:1319–1324.
- Macrez R, Ali C, Toutirais O, Le Mauff B, Defer G, Dirnagl U, Vivien D. 2011. Stroke and the immune system: from pathophysiology to new therapeutic strategies. Lancet Neurol. 10:471–480.
- Matsui Y, Kyoi S, Takagi H, Hsu CP, Hariharan N, Ago T, Vatner SF, Sadoshima J. 2008. Molecular mechanisms and physiological significance of autophagy during myocardial ischemia and reperfusion. Autophagy. 4:409–415.
- McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. 2005. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 280:20493–20502.
- Mizushima N, Yoshimori T, Levine B. 2010. Methods in mammalian autophagy research. Cell. 140:313–326.
- Nam HG, Kim W, Yoo DY, Choi JH, Won MH, Hwang IK, Jeong JH, Hwang HS, Moon SM. 2013. Chronological changes and effects of AMP-activated kinase in the hippocampal CA1 region after transient forebrain ischemia in gerbils. Neurol Res. 35:395–405.
- Ozbal S, Erbil G, Kocdor H, Tuğyan K, Pekçetin C, Ozoğul C. 2008. The effects of selenium against cerebral ischemia-reperfusion injury in rats. Neurosci Lett. 438:265–269.
- Powers WJ, Derdeyn CP, Biller J, Coffey CS, Hoh BL, Jauch EC, Johnston KC, Johnston SC, Khalessi AA, Kidwell CS, et al. 2015. 2015 American Heart Association/American Stroke Association focused update of the 2013 guidelines for the early management of patients with acute ischemic stroke regarding endovascular treatment: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 46:3020–3035.
- Pamenter ME, Perkins GA, McGinness AK, Gu XQ, Ellisman MH, Haddad GG. 2012. Autophagy and apoptosis are differentially induced in neurons and astrocytes treated with an in vitro mimic of the ischemic penumbra. PLoS One. 7:e51469.
- Park JS, Manzanero S, Chang JW, Choi Y, Baik SH, Cheng YL, Li YI, Gwon AR, Woo HN, Jang J. 2012. Calsenilin contributes to neuronal cell death in ischemic stroke. Brain Pathol. 23:402–412.
- Puyal J, Clarke PG. 2009. Targeting autophagy to prevent neonatal stroke damage. Autophagy. 5:1060–1061.
- Rami A, Langhagen A, Steiger S. 2008. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol Dis. 29:132–141.
- Reggiori F, Klionsky DJ. 2002. Autophagy in the eukaryotic cell. Eukaryotic Cell. 1:11–21.
- Sheng R, Zhang LS, Han R, Liu XQ, Gao B, Qin ZH. 2010. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy. 6:482–494.
- Szydlowska K, Tymianski M. 2010. Calcium, ischemia and excitotoxicity. Cell Calcium. 47:122–129.
- Shen K, Wang Y, Zhang Y, Zhou H, Song Y, Cao Z, Kou J, Yu B. 2015. Cocktail of four active components derived from Sheng Mai San inhibits hydrogen peroxide-induced PC12 cell apoptosis linked with the caspase-3/ROCK1/MLC pathway. Rejuvenation Res. 18:517–527.
- Tian F, Deguchi K, Yamashita T, Ohta Y, Morimoto N, Shang J, Zhang X, Liu N, Ikeda Y, Matsuura T, et al. 2010. In vivo imaging of autophagy in a mouse stroke model. Autophagy. 6:1107–1114.
- Wang XM, Zhao C, Wang SX, Lin XY, Liu XL. 2015. Research on Xueshuantong injection through blood brain barrier. Drug Eval Res. 38:384–389.
- Wang N, Minatoguchi S, Arai M, Uno Y, Nishida Y, Hashimoto K, Xue-Hai C, Fukuda K, Akao S, Takemura G, et al. 2002. Sheng-Mai-San is protective against post-ischemic myocardial dysfunction in rats through its opening of the mitochondrial KATP channels. Circ J. 66:763–768.
- Wei X, Ye X, Xie Y, Zou Y, Zhao X, Han J, Wang X, Ma Y, Bi Q, Xie Q, et al. 2011. Post-marketed re-evaluation of fleabane injection and Dengzhan Shengmai capsule study on treatment in patients with ischemic stroke. Zhongguo Zhong Yao Za Zhi. 36:2789–2792.
- Wei Y, Pattingre S, Sinha S, Bsaaik M, Levine B. 2008. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 30:678–688.
- Winters L, Winters T, Gorup D, Mitrečić D, Curlin M, Križ J, Gajovic S. 2013. Expression analysis of genes involved in TLR2-related signaling pathway: Inflammation and apoptosis after ischemic brain injury. Neuroscience. 238:87–96.
- Wirawan E, Lippens S, Vanden Berghe T, Romagnoli A, Fimia GM, Piacentini M, Vandenabeele P. 2012. Beclin1: a role in membrane dynamics and beyond. Autophagy. 8:6–17.
- Wong CH, Crack PJ. 2008. Modulation of neuro-inflammation and vascular response by oxidative stress following cerebral ischemia-reperfusion injury. Curr Med Chem. 15:1–14.
- Xie R, Wang P, Ji X, Zhao H. 2013. Ischemic post-conditioning facilitates brain recovery after stroke by promoting Akt/mTOR activity in nude rats. J Neurochem. 127:723–732.
- Xu M, Zhang HL. 2011. Death and survival of neuronal and astrocytic cells in ischemic brain injury: a role of autophagy. Acta Pharmacol Sin. 32:1089–1099.
- Xu BB, Liu CQ, Gao X, Zhang WQ, Wang SW, Cao YL. 2005. Possible mechanisms of the protection of ginsenoside Re against MPTP-induced apoptosis in substantia nigra neurons of Parkinson's disease mouse model. J Asian Nat Prod Res. 7:215–224.
- Yan W, Zhang H, Bai X, Liu Y, Dong H, Xiong L. 2011. Autophagy activation is involved in neuroprotection induced by hyperbaric oxygen preconditioning against focal cerebral ischemia in rats. Brain Res. 1402:109–121.
- Yang HP, Li L, Chai CZ, Kou JP, Yu BY, Yan YQ. 2013. Neuroprotective effect of Shengmai Injection on expression of tissue factor and related signal pathways in mice with cerebral ischemia- reperfusion injury. Chin J Exp Trad Med Formulae. 19:191–196.
- Yang Z, Klionsky DJ. 2010. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 22:124–131.
- Yuan QL, Yang CX, Xu P, Gao XQ, Deng L, Chen P, Sun ZL, Chen QY. 2007. Neuroprotective effects of ginsenoside Rb1 on transient cerebral ischemia in rats. Brain Res. 1167:1–12.
- Yousuf S, Atif F, Ahmad M, Hoda N, Ishrat T, Khan B, Islam F, et al. 2009. Resveratrol exerts its neuroprotective effect by modulating mitochondrial dysfunctions and associated cell death during cerebral ischemia. Brain Res. 1250:242–253.
- Zhang F, Yin W, Chen J. 2004. Apoptosis in cerebral ischemia: executional and regulatory signaling mechanisms. Neurol Res. 26:835–845.
- Zheng C, Han J, Xia W, Shi S, Liu J, Ying W. 2012. NAD(+) administration decreases ischemic brain damage partially by blocking autophagy in a mouse model of brain ischemia. Neurosci Lett. 512:67–71.