Abstract
Objectives. Through the hexosamine biosynthetic pathway (HBP) proteins are modified by O-linked-β-N-acetylglucosamine (O-GlcNAc), which acts as a stress sensor. Augmentation of O-GlcNAc confers cardioprotection against ischemia- reperfusion injury, but its role in ischemic preconditioning (IPC) is unknown. Azaserine and alloxan are unspecific blockers of the HBP and have been used to block the cardioprotective effects of O-GlcNAc. We hypothesized that IPC reduces infarct size and increases O-GlcNAc levels in hearts subjected to ischemia-reperfusion injury, and that these effects could be blocked by azaserine and alloxan. Design. Isolated rat hearts subjected to 40 min global ischemia and 120 min reperfusion were randomized to control, IPC, IPC + azaserine or alloxan, or control + azaserine or alloxan. The effects on infarct size, hemodynamic recovery, myocardial O-GlcNAc levels, and HBP enzyme activities were determined. Results. IPC reduced infarct size, increased O-GlcNAc levels, O-GlcNAc-transferase levels, and O-GlcNAc-transferase activity. Azaserine and alloxan did not block the effect of IPC on O-GlcNAc levels and O-GlcNAc-transferase activity. Conclusions. IPC increased O-GlcNAc levels though increased O-GlcNAc-transferase expression and activity. Azaserine and alloxan failed to block these effects presumably due to poor specificity and sensitivity of the blockers, and IPC-mediated cardioprotection may therefore still be dependent on O-GlcNAc.
Introduction
Posttranslational modification of nuclear, cytoplasmic, and mitochondrial proteins by O-linked attachment of β-N-acetylglucosamine (O-GlcNAc) is essential in numerous biological processes and important in signal transduction (Citation1). O-GlcNAc acts as a stress-induced signal with pro-survival effects (Citation2). Ischemic preconditioning (IPC), a novel cardioprotective mechanism where short periods of sublethal ischemia followed by reperfusion exerts a protective effect against subsequent lethal ischemia (Citation3), increases O-GlcNAc levels (Citation4), but it has not been determined whether IPC relies on O-GlcNAc for cardioprotection.
O-GlcNAc is one endpoint of the hexoamine biosynthesis pathway (HBP), where fructose-6-phosphate is metabolized to glucosamine-6-phosphate by the enzyme L-glutamine-D-fructose-6-phosphate amidotransferase (GFAT). Through several enzymatic steps glucosamine-6-phosphate is converted into uridine-diphosphate-N-acetylglucosamine (UDP-GlcNAc), which acts as substrate for the enzyme uridine diphospho-N-acetylglucosamine:polypeptide-N-acetylglucosaminyltransferase (O-GlcNAc transferase) that catalyzes the addition of O-GlcNAc to serine and threonine residues. The removal of O-GlcNAc is catalyzed by β-hexoamininidase (O-GlcNAcase). Several inhibitors of HBP are available. The GFAT inhibitor azaserine and the O-GlcNAc transferase inhibitor alloxan have successfully been used to block glucosamine-mediated increase in O-GlcNAc levels in isolated perfused hearts, but both inhibitors lack sensitivity and specificity (Citation5,Citation6).
The aim of the present study was to investigate whether cardioprotection by IPC is dependent of O-GlcNAcylation of myocardial proteins, and the role of O-GlcNAcase and O-GlcNAc transferase activity for augmentation of O-GlcNAc by IPC. We hypothesized that the GFAT inhibitor azaserine and the O-GlcNAc transferase inhibitor alloxan would block IPC-induced cardioprotection and O-GlcNAcylation of myocardial proteins.
Methods
Ethical approval
The study conforms to Danish law for animal research (Act no. 1306 of 23/11/2007 Rules in force, The Danish Ministry of Justice) and is in accordance with guidelines for animal care and use approved by The Animal Experiments Inspectorate in Denmark.
Experimental groups
Infarct study: Isolated perfused heart experiments were performed on male Wistar rats (M&B, Taconic, Ry, Denmark), weighing 280–320 g, which were randomized to the following experimental groups: (i) control; (ii) IPC; (iii) control + 80 μM azaserine; (iv) IPC + 40 μM azaserine; (v) IPC + 80 μM azaserine; (vi) IPC + 120 μM azaserine; (vii) IPC + 160 μM azaserine; (viii) control + 5 mM alloxan; and (ix) IPC + 5 mM alloxan.
Isolated perfused rat hearts were subjected to 50 min of stabilization, 40 min of global no-flow ischemia, and 120 min of reperfusion. Hearts in the IPC groups were subjected to 2 × 5 min of global no-flow ischemia with 5 min reperfusion in between prior to index ischemia, while control hearts were subjected to time-matched standard perfusion (). Experimental groups were subjected to azaserine or alloxan, and these agents were added to the perfusion buffer 15 min before the IPC stimulus or time-matched in control groups and were present throughout the rest of the experiment.
Figure 1. Experimental groups and isolated heart perfusion protocols. All groups were TTC stained for infarct size determination. Tissue analysis for characterization of HPB were performed on selected groups.
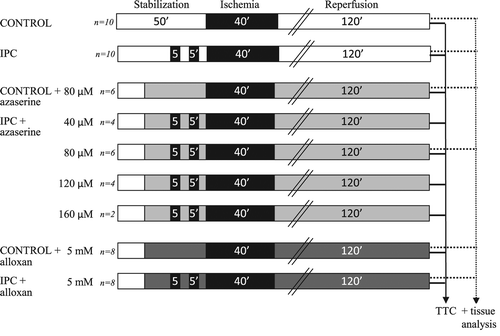
O-GlcNAc study: In selected groups of the isolated perfused rat hearts (), a piece of myocardium was snap frozen at the end of reperfusion for determination of O-GlcNAc levels and characterization of HBP.
The isolated perfused heart
Rats were anaesthetized with a mixture of 0.25 mg/kg bodyweight Dormicum® (midazolam; Roche, Basel, Switzerland) and 0.5 mg/kg bodyweight Hypnorm® (fentanyl citrate 0.315 mg/mL + Fluanison 10 mg/mL; Janssen, Birkerød, Denmark) and connected to a ventilator. Through laparotomy and thoracotomy the heart was rapidly dissected free from surrounding structures, and the animal was heparinized by injection of a 1,000 IU/kg heparin bolus through the femoral vein. The aorta cannula was placed in situ in ascending aorta and retrograde perfusion with 37°C Krebs–Henseleit buffer (containing (in mmol/L) NaCl2 (118), KCl (4.8), NaHCO3 (27.2), MgCl2 (1.2), KH2PO4 (1.0) CaCl2 (2.0), glucose (10)) was established in situ. The heart was rapidly excised and mounted in the Langendorff apparatus, submerged in 37°C Krebs–Henseleit buffer and perfused at constant pressure of 80 mm Hg. Recirculating Krebs–Henseleit buffer was continuously oxygenated with 95% O2 and 5% CO2 to maintain a pH of 7.35–7.45.
The left atrial auricle was cut off and a balloon catheter (size 7, Hugo Sachs Electronics, March-Hugstetten, Germany), connected to a pressure transducer, was instrumented into the left ventricular, for continuous hemodynamic measurements. Diastolic pressure was set to 7–10 mmHg at the beginning of the experiment. Flow was continuously measured by a flowmeter (Hugo Sachs Electronics, March-Hugstetten, Germany). Data were acquired and analyzed using a dedicated core software platform (Notocord Hem evolution, Croissy sur Seine, France.)
Determination of infarct size
At the end of reperfusion a 1.5 mm slice of the heart was immediately snap frozen in liquid nitrogen and stored at − 80°C for later western blotting and enzyme activity analysis. The rest of the heart has frozen at − 80°C for 15 min and then cut into 1.5 mm slices and immersed in 1% 2,3,5-triphenyltetrazolium chloride (Sigma, St.Louis, Mo, USA) at 37°C for 3 min at pH = 7.4. Hearts were stored overnight in 4% formaldehyde buffer. On the following day each heart slice was weighed and scanned with a flatbed scanner (HP ScanJet 4300C, Hewlett Packard, Palo Alto, CA, USA). Area of left ventricle (LV), which corresponds to the area at risk (AAR), and the area of infarction (IS) were assessed by observer delineation of the basal side of the heart slice using computer planimetry (UTHSCSA Image Tool for Windows (version 3.0), San Antonio, TX, USA). Infarct size of the snap frozen slice was determined by delineation of the apex side of the slice above. Infarct size was expressed as a percentage IS of AAR. Measurements were weighted with the weight of each individual slice. All analyses were performed in a blinded manner.
O-GlcNAc analysis
Tissue samples were thawed in extraction buffer (15 mM HEPES pH 7.3, 10% v/v glycerol, 0.5% v/v NP-40, 250 mM NaCl, 1 mM EDTA, 0.1 mM proteases inhibitor PMSF, 10 mM phosphatases inhibitor Potassium Fluoride, 3 μM deacetylation inhibitor Trichostatin A, 10 mM phosphatases inhibitor β-glycerophosphate, PIC 2 and PIC 3 (proteases inhibitor Cocktail, Roche), and 2 μM Thiamet G, an inhibitor of O-GlcNAcase), grounded with an electric grinder, and spun 18,000 × g at 4°C for 30 min. Total cell lysates (TCL) were transferred to new tubes and protein concentrations were determined using Pierce 660 nm Protein Assay (Thermo Scientific). Protein of 20 μg was loaded on four 8% PAGE gels (Bis-Tris, Invitrogen). Gels were transferred to nitrocellulose membrane, blocked in 3% w/v milk in TBST for 1 hr, and primed with antibodies: anti- O-GlcNAc antibody (CDT 110.6, Gift – CoreC4), anti-O-GlcNAc antibody (CTD 110.6) + 100 mM GlcNAc (Sigma), anti-Hex C (345, Gift – CoreC4), anti-OGT (DM17, Sigma), and anti-actin antibody (Sigma). Densitometry was performed on specific bands and for O-GlcNAc on entire lane of each sample using software (UTHSCSA Image Tool for Windows (version 3.0), San Antonio, TX, USA). All densitometry were calculated relative to corresponding actin blot. Values were presented as percentage of control (set at 100%).
Hexoamin biosyntethic pathway enzyme activity analysis
Freshly extracted total cell lysate (TCL) was desalted into OGT desalting buffer (20 mM Tris, HCl pH7.8, 20% v/v glycerol, and 0.02% w/v Sodium azide) using Zebra Spin Desalting Plate (Thermo scientific). Protein concentrations were estimated using Pierce 660 nm Protein Assay (Thermo Scientific).
O-GlcNAcase activity was estimated by measuring the cleavage rate of a synthetic substrate, p-nitrophenol N-acetylglucosamine (pNP-GlcNAc). The activity assay was performed on 50 μg protein in 100 μl of 50 mM sodium cacodylate, 2 mM pNP-GlcNAc, 0.5 mg/ml of bovine serum albumin and 50 mM GalNAc at 37°C for 30, and 60 min for each sample. The reactions were stopped with addition of 100 μl sodium carbonate. Absorbance was measured at 400 nm. Activity is reported as picomoles of cleavage per min per mg of cell extract.
O-GlcNAc transferase activity was estimated by determining the rate at which [H3]-UDP-N-acetyl-D-glucosamine (Perkin Elmer) was transferred to an acceptor peptide, casein kinase II peptide. Assays (10 μg of desalted cell extract) were performed in a final volume of 50 μl, containing 50 mM sodium cacodylate, 0.1 mg/ml of bovine serum albumin, and 2.5 mM AMP to competitively inhibit any pyrophospatase, 0.25 unit of calf intestinal alkaline phosphatase, 0.5 μCi of UDP-[3H]GlcNAc (Specific Activity 60 μCi/mole), and 1 mM CKII peptide substrate (PGGSTPVSSANMM). Reactions were performed at room temperature for 30 and 60 min for each sample and stopped by adding 50 mM formic acid. The reactions were purified over C18 cartridge (Phenomenex), eluted with methanol into scintillation fluid, and counted. Activity was reported as picomoles incorporated O-GlcNAc per min per mg of cell extract.
Statistical analysis
All values presented were mean ± SEM. Data were analyzed by one-way ANOVA and two-way ANOVA with repeating measurements with post hoc analysis (Bonferroni) as appropriate. Correlation was assessed using Pearson's correlation. Statistical significance was accepted as p-value < 0.05, two-tailed.
Results
Infarct size and hemodynamic recovery
IPC reduced infarct size (16 ± 4% vs 41 ± 4%; p < 0.01) () and improved postischemic recovery of LVDP, RPP, dP/dtmax, and dP/dtmin. (p < 0.05 for all measurements) (). In the azaserine dose-response experiment, infarct size in the IPC groups treated with azaserine was lower than infarct size in the nontreated control group (40 μM: 15 ± 3%; 80 μM: 19 ± 5%; 120 μM: 14 ± 5%; and 160 μM: 8 ± 2% vs nontreated control: 41 ± 4% of LV p < 0.05 for all comparisons). IPC + 80 μM azaserine had the largest infarct size of the azaserine IPC groups and only this concentration was tested on a control group. IPC + 80 μM azaserine had smaller infarct size compared to the control group + 80 μM azaserine (), even though this difference was not statistically significant (p = 0.08). Azaserine of 80 μM abolished the effect of IPC on RPP, dP/dtmax, dP/dtmin compared to control + 80 μM azaserine (p = 0.2, p = 0.2, and p = 0.6, respectively).
Figure 3. a: Infarct size in control, IPC, control + 80 μM azaserine, IPC + 80 μM azaserine, control + 5 mM alloxan, and IPC + 5 mM alloxan treated hearts. b: O-GlcNAc levels in in control, IPC, control + 80 μM azaserine, IPC + 80 μM azaserine, control + 5 mM alloxan, and IPC + 5 mM alloxan treated hearts. c: O-GlcNAc transferase activity in control, IPC, control + 80 μM azaserine, IPC + 80 μM azaserine, control + 5 mM alloxan, and IPC + 5 mM alloxan treated hearts. d: O-GlcNAc transferase levels in control, IPC, control + 80 μM azaserine, IPC + 80 μM azaserine, control + 5 mM alloxan, and IPC + 5 mM alloxan treated hearts. e: O-GlcNAcase activity in control, IPC, control + 80 μM azaserine, IPC + 80 μM azaserine, control + 5 mM alloxan, and IPC + 5 mM alloxan treated hearts. f: O-GlcNAcase levels in control, IPC, control + 80 μM azaserine, IPC + 80 μM azaserine, control + 5 mM alloxan, and IPC + 5 mM alloxan treated hearts. All data presented are mean ± SEM.
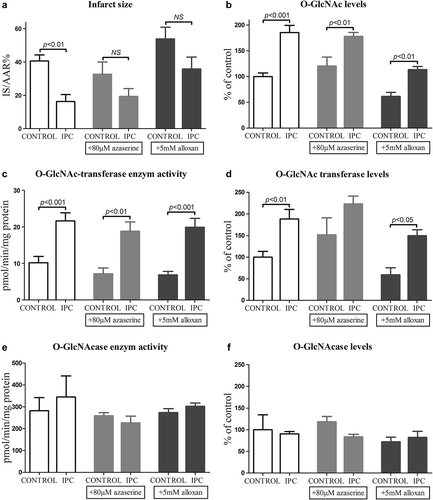
Figure 4. Left ventricular developed pressure (LVDP) during reperfusion in a: control and IPC hearts, b: control + 80 μM azaserine and IPC + 80 M azaserine treated hearts, and c: control + 5 mM alloxan and IPC + 5 mM alloxan. All data presented are mean ± SEM. **p < 0.05 compared to control.
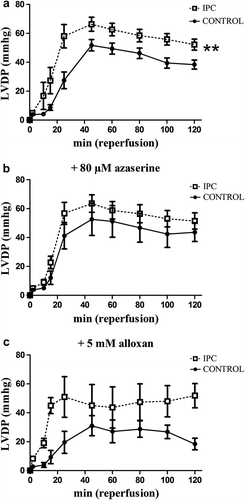
The difference in infarct size () and hemodynamic recovery () between control and IPC was no longer statistically significant after addition of alloxan, suggesting that alloxan attenuated the effect of IPC. But hearts treated with IPC + 5 mM alloxan had improved postischemic RPP, dP/dtmax, and dP/dtmin compared to control + 5 mM alloxan (p = 0.037, p = 0.01, and p = 0.01, respectively) (data not shown).
The infarct size in the control + 5 mM alloxan was larger than the untreated control group and control + 80 μM azaserine group, but this difference was not statistically significant (41 ± 4% and 33 ± 7%, and 54 ± 7%; p = 0.07).
Characterization of HBP
IPC increased O-GlcNAc levels compared to control (190 ± 10% vs 100 ± 7%; p < 0.0001), increased O-GlcNAc transferase levels (190 ± 20% vs 100 ± 10% p < 0.001), but had no effect of O-GlcNAcase levels (90 ± 50% and 100 ± 30%; p = 0.5) ( and ). Similarly IPC increased O-GlcNAc transferase activity compared to control (22 ± 2 2 pmol/min/mg protein vs 10 ± 2 pmol/min/mg protein; p < 0.001), but IPC had no effect on O-GlcNAcase activity (345 ± 96 pmol/min/mg protein and 282 ± 61 pmol/min/mg protein p > 0.05) ().
Figure 2. Representative O-GlcNAc (CTD 110.6) and actin immunoblots from control, IPC, control + 80 μM azaserine, IPC + 80 μM azaserine, control + 5 mM alloxan, and IPC + 5 mM alloxan.
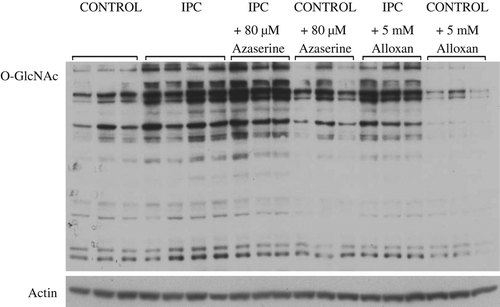
Azaserine and alloxan did not block the effect of IPC on O-GlcNAc, O-GlcNAc transferase, or O-GlcNAcase levels, and O-GlcNAc transferase, and O-GlcNAcase activities ( and ). However, it is evident that changes in O-GlcNAc levels and O-GlcNAc transferase activity and levels change in parallel and that increases in these variables are associated with parallel decreases in infarct size. On an individual basis we did not find significant inverse correlations between O-GlcNAc levels and infarct size (data not shown).
Discussion
The present study demonstrates that IPC augments O-GlcNAc by increased O-GlcNAc transferase activity. The O-GlcNAc transferase blocker alloxan and the GFAT blocker azaserine did not sufficiently block the cardioprotective effect, the increase in myocardial O-GlcNAc content, or O-GlcNAc transferase activity.
The study is confirmatory by showing that IPC increases O-GlcNAc levels. We identified that this is mediated by an increased O-GlcNAc transferase activity, due to increased O-GlcNAc transferase expression. Both alloxan and azaserine failed to completely block the increase in myocardial O-GlcNAc levels and O-GlcNAc transferase activity. This is presumably due to insufficient specificity of these blockers. This finding complicates any conclusion about the dependency of O-GlcNAc on the cardioprotective effect of IPC. However, it is evident from that attenuation of the increment of myocardial O-GlcNAc levels by alloxan is reflected in a reduction in infarct size in the alloxan treated control and IPC groups, while increase in O-GlcNAc levels by azaserine is reflected as an increase in infarct size in the azaserine-treated control and IPC groups.
The reduction of infarct size and improved hemodynamic recovery by IPC were not statistically significant in hearts treated with azaserine when compared to the azaserine-treated control group, whereas the increase in O-GlcNAc was significant. It is obvious that the difference is not different from the untreated group and the lack of statistical significance is merely due to increased variation in these study groups. Azaserine and alloxan obviously did not block the HBP and O-GlcNAcylation of proteins to a very high degree in this study. All together the findings of the study suggest that augmentation in O-GlcNAc levels is involved in the mechanism of cardioprotection by IPC, and alloxan and azaserine are insufficient blockers of the HBP in an isolated perfused heart model.
Alloxan is a very unstable compound with a half-life of 1.5 min (Citation7) and the concentration may not have been high enough when used in a recirculation model in the Langendorff model. Furthermore, both azaserine and alloxan are very weak and not specific blockers of GFAT and O-GlcNAc transferase (Citation5,Citation6,Citation8). Other blockers of the HBP are available, but also these have demonstrated variable efficiency in different laboratories (Citation9). We have no specific explanation for our lack of success with azaserine and alloxan in the isolated perfused heart model, while others have (Citation10,Citation11).
Alloxan is a validated O-GlcNAc transferase blocker in isolated cells (Citation12), and we demonstrated by measuring O-GlcNAc transferase activity that alloxan failed to block O-GlcNAc transferase in our isolated rat heart model. We did not measure GFAT activity in order to investigate if azaserine indeed failed to block GFAT or if the evident failure to block increase in O-GlcNAc was due to an increase in substrates downstream from GFAT in the HBP, like glucosamine or N-acetylglucosamine (Citation13). We can therefore not conclude if increase in O-GlcNAc is involved in mediating cardioprotection by IPC. This is a limitation of our study.
In accordance with our study, others have demonstrated that IPC augments O-GlcNAc levels (Citation4). Regardless of the means of increasing cardiac O-GlcNAc levels, for example, by inhibition of O-GlcNAcase (Citation14), overexpression of O-GlcNAc transferase (Citation15,Citation16), or perfusion with glutamine (Citation10,Citation11), any increment reduces injury following ischemia-reperfusion injury. Similarly to IPC (Citation17,Citation18), cardioprotection by augmentation of O-GlcNac levels is mediated through inhibition of mPTP opening (Citation4).
In conclusion, IPC increased O-GlcNAc levels through increased O-GlcNAc-transferase expression, and infarct size after ischemia-reperfusion injury is reflected in O-GlcNAc levels. Azaserine and alloxan did not block these effects presumably due to insufficient specificity and sensitivity of the blockers. Studies with specific blockers of the HBP are needed to determine the involvements of O-GlcNAc on cardioprotection by IPC.
Acknowledgements
We wish to thank the Core C4 (NHLBI P01HL107153) at the Johns Hopkins University School of Medicine for the gift of O-GlcNAcase antibody (345; G.W. Hart) and O-GlcNAc antibody (Natasha Zachara).
Funding
This work was supported by the Danish Research Council (11-108354), The Danish Strategic Research Council (11-1115818), and Institute of Clinical Medicine, Aarhus University, and American Heart Association (SD930162N to N.E.Z) and the National Heart Lung and Blood Institute (R21-HL-108003 and PO1HL107153 to N.E.Z.)
Declaration of interest: The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
We greatly appreciate the technical assistance from Anja Helveg Larsen and Casper Elkjær.
References
- Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825–58.
- Zachara NE, O’Donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J Biol Chem 2004;279:30133–42.
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986;74:1124–36.
- Jones SP, Zachara NE, Ngoh GA, Hill BG, Teshima Y, Bhatnagar A, et al. Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation 2008;117:1172–82.
- Lyons SD, Sant ME, Christopherson RI. Cytotoxic mechanisms of glutamine antagonists in mouse L1210 leukemia. J Biol Chem 1990;265:11377–81.
- Suzuki H, Kumagai H, Tochikura T. Gamma-Glutamyltranspeptidase from Escherichia coli K-12: purification and properties. J Bacteriol 1986;168:1325–31.
- Lenzen S, Munday R. Thiol-group reactivity, hydrophilicity and stability of alloxan, its reduction products and its N-methyl derivatives and a comparison with ninhydrin. Biochem Pharmacol 1991;42:1385–91.
- Dorfmueller HC, Borodkin VS, Blair DE, Pathak S, Navratilova I, van Aalten DM. Substrate and product analogues as human O-GlcNAc transferase inhibitors. Amino Acids 2011;40:781–92.
- Zachara NE. The roles of O-linked beta-N-acetylglucosamine in cardiovascular physiology and disease. Am J Physiol Heart Circ Physiol 2012;302:H1905–H1918.
- Liu J, Marchase RB, Chatham JC. Glutamine-induced protection of isolated rat heart from ischemia/reperfusion injury is mediated via the hexosamine biosynthesis pathway and increased protein O-GlcNAc levels. J Mol Cell Cardiol 2007;42:177–85.
- Liu J, Marchase RB, Chatham JC. Increased O-GlcNAc levels during reperfusion lead to improved functional recovery and reduced calpain proteolysis. Am J Physiol Heart Circ Physiol 2007;293:H1391–H1399.
- Konrad RJ, Zhang F, Hale JE, Knierman MD, Becker GW, Kudlow JE. Alloxan is an inhibitor of the enzyme O-linked N-acetylglucosamine transferase. Biochem Biophys Res Commun 2002;293:207–12.
- Butkinaree C, Park K, Hart GW. O-linked beta-N-acetylglucosamine (O-GlcNAc): extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim Biophys Acta 2010;1800: 96–106.
- Laczy B, Marsh SA, Brocks CA, Wittmann I, Chatham JC. Inhibition of O-GlcNAcase in perfused rat hearts by NAG-thiazolines at the time of reperfusion is cardioprotective in an O-GlcNAc-dependent manner. Am J Physiol Heart Circ Physiol 2010;299:H1715–H1727.
- Champattanachai V, Marchase RB, Chatham JC. Glucosamine protects neonatal cardiomyocytes from ischemia-reperfusion injury via increased protein O-GlcNAc and increased mitochondrial Bcl-2. Am J Physiol Cell Physiol 2008;294:C1509–C1520.
- Ngoh GA, Watson LJ, Facundo HT, Dillmann W, Jones SP. Non-canonical glycosyltransferase modulates post-hypoxic cardiac myocyte death and mitochondrial permeability transition. J Mol Cell Cardiol 2008;45:313–25.
- Argaud L, Gateau-Roesch O, Chalabreysse L, Gomez L, Loufouat J, Thivolet-Bejui F, et al. Preconditioning delays Ca2+-induced mitochondrial permeability transition. Cardiovasc Res 2004;61:115–22.
- Javadov SA, Clarke S, Das M, Griffiths EJ, Lim KH, Halestrap AP. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J Physiol 2003;549:513–24.