
Abstract
Objectives. To investigate survival, treatment escalation, effects of first-line single- and first-line combination therapy and prognostic markers in idiopathic- (IPAH), hereditary- (HPAH) and connective tissue disease-associated (CTD-PAH) pulmonary arterial hypertension (PAH). Design. Retrospective analysis of medical journals from PAH patients at Skåne University Hospital 2000–2011. Results. 1-, 2- and 3-year survival was 87%, 67%, and 54%, respectively, for the entire population, but worse (p = 0.003) in CTD-PAH than IPAH/HPAH. After 1, 2 and 3 years, 58%, 41% and 24% of patients starting on single therapy were alive on single therapy. 37.5% of patients on first-line single therapy received escalated treatment at first follow-up. First-line combination therapy more greatly decreased pulmonary vascular resistance index (PVRI, p = 0.017) than first-line single therapy. Only first-line combination therapy improved (p = 0.042) cardiac index (CI). Higher mean right atrial pressure (MRAP, p = 0.018), MRAP/CI (p = 0.021) and WHO functional class (p < 0.001) and lower 6-min walking distance (6MWD, p = 0.001) at baseline, and higher PVRI (p = 0.008) and lower 6MWD (p = 0.004) at follow-up were associated with worse outcome. Conclusions. We confirm improved survival with PAH-targeted therapies. Survival is still poor and early treatment escalation frequently needed. First-line combination therapy may more potently improve haemodynamics. MRAP/CI may represent a new prognostic marker in PAH.
Introduction
Pulmonary arterial hypertension (PAH) is a disease of distal pulmonary arteries characterized by medial hypertrophy, intimal proliferation and fibrosis, adventitial thickening, thrombosis, and complex lesions, which ultimately leads to right ventricular failure and death (Citation1,Citation2). Pulmonary hypertension is defined by a mean pulmonary arterial pressure (MPAP) of 25 mmHg or higher at rest and the diagnosis of PAH is set after excluding other reasons of increased pulmonary arterial blood pressure, such as left heart disease, lung disease or hypoxaemia, and chronic thromboembolism (Citation3). PAH may be idiopathic (IPAH), hereditary (HPAH) or associated to other conditions, including connective tissue diseases (CTD-PAH), congenital heart diseases, portal hypertension, HIV- or schistosomiasis infection and use of anorexigens (Citation4).
Survival is still poor in PAH, although it has improved since the introduction of PAH-targeted therapies, as compared with untreated patients from a large registry from the 1980s (Citation5–8). The current PAH-targeted therapies include oral single- or double endothelin receptor antagonists (ERA) ambrisentan and bosentan, phosphodiesterase-5 inhibitors (PDE5i) sildenafil and tadalafil, and inhalation, subcutaneous or intravenous prostacyclin analogues iloprost, treprostinil and epoprostenol (Citation9). At present, a new per oral ERA, macitentan (Citation10), and a per oral soluble guanylate cyclase stimulator, riociguat (Citation11), are being introduced for the treatment of PAH. A subgroup of patients with better prognosis, defined as acute vasoresponders, is treated with calcium channel blockers (CCB) (Citation12,Citation13). Despite these new treatments, lung transplantation remains as a last treatment resort (Citation3).
The European Society of Cardiology's guidelines recommend at present a ‘treat-to-target’ approach based on first-line single therapy, with sequential addition of drugs at follow-up if prognostic variables are unstable and dissatisfactory (Citation3). Nickel et al. recently showed that IPAH patients who reach treatment goals have a better prognosis (Citation14). Our clinical experience indicates that early introduction of combination therapy is often necessary and that patients may profit from combination therapy already at diagnosis. However, only sparse evidence exists for the benefit of first-line combination therapy (Citation15,Citation16). The identification of new prognostic markers at diagnosis and at follow-up could aid in identifying patients in early need of more aggressive treatment.
With this retrospective study, our aims were to in a ‘real-life’, single-centre setting; estimate survival and frequency of treatment escalation, describe and compare the haemodynamic effects of first-line single- and first-line combination therapy, respectively, in patients with IPAH, HPAH and CTD-PAH, and identify prognostic markers at baseline and follow-up. We hypothesized that survival has improved since introduction of PAH-targeted therapies that many patients need early treatment escalation and that first-line combination therapy may more rapidly and potently than first-line single therapy improve haemodynamics.
Material and methods
The study was approved by the local ethics board in Lund (Dnr 2011/777, Dnr 2011/368, Dnr 2010/114) and adhere to the ethical standards laid down in the Declaration of Helsinki.
Medical journal data was gathered from patients above 18 years of age diagnosed with IPAH, HPAH or CTD-PAH at Skåne University Hospital in Lund, Sweden, from 1 January 2000 to 31 December 2010 and devoid of multiple PH diagnoses. The data were subsequently included in the Lund cohort of the Swedish Pulmonary Arterial Hypertension Register (SPAHR). IPAH- and HPAH patients were treated as an entity. Patients were followed until 31 December 2011 or death.
Right-heart catheterizations (RHC) were performed at the Haemodynamic Laboratory at Skåne University Hospital in Lund using a Swan-Ganz catheter, preferentially inserted via the right internal jugular vein. MPAP, mean right-atrial pressure (MRAP), pulmonary capillary wedge pressure (PCWP) and cardiac output (CO), measured by thermodilution, were registered at the time of RHC. Cardiac index (CI) = CO/body surface area, transpulmonary gradient (TPG) = MPAP − PCWP, diastolic pulmonary vascular pressure gradient (DPG) = diastolic PAP − PCWP, pulmonary vascular resistance index (PVRI) = TPG/CI, right ventricular stroke work index (RVSWI) = stroke volume/body surface area ·(MPAP − MRAP) · 0.0136 were calculated. MRAP/CI was calculated for patients with MRAP values more than 0 mmHg. The 6-min walking test was performed in accordance with the American Thoracic Society's guidelines (Citation17).
Treatment decisions at diagnosis and at follow-up were made by experienced cardiologists in collaboration with rheumatologists with a special interest in PAH. Decisions were based on a combination of clinical indices, including haemodynamics, functional class, 6-min walking distance (6MWD), echocardiography, electrocardiogram, ventilation/perfusion lung scan, magnetic resonance imaging of the heart, high-resolution computed tomography of the thorax and NT-proBNP levels, in relation to available guidelines and recommendations at the time.
Survival and treatment escalation
1-, 2- and 3-year survival rates were estimated for all patients with IPAH/HPAH (n = 39) or CTD-PAH (n = 38), respectively. Patients were censored at transplantation or 31 December 2011.
Survival rates were compared for IPAH/HPAH and CTD-PAH patients, respectively, and for patients that received first-line single- or first-line combination therapy, respectively.
The proportion of patients that started on first-line single therapy and still were alive on single therapy after 1-, 2- and 3-years, that is, time to death, transplantation or escalation to combination therapy, was estimated. Patients were censored 31 December 2011. The frequency of treatment escalation from first-line single therapy to combination therapy at first RHC follow-up was investigated. Haemodynamic parameters were compared at baseline and first RHC follow-up for patients who received first-line single therapy and who had, or had not, been escalated to combination therapy at first RHC follow-up. The frequency of lung transplantations and survival after lung transplantation were furthermore investigated.
Five patients had already started PAH-targeted therapies before first RHC had been performed at our centre. Three of these patients (one IPAH/HPAH, 2 CTD-PAH) had started treatment based on echocardiographic diagnosis of pulmonary hypertension. One patient (CTD-PAH) had started treatment in relation to a clinical trial and one patient (IPAH/HPAH) was first diagnosed with pulmonary hypertension at another hospital in the 1990s. For these patients overall survival and survival on first-line single therapy were estimated from date of first RHC at our centre, except for the IPAH/HPAH patient diagnosed at another hospital in the 1990s. For all other patients, survival rates were estimated from time of PAH diagnosis when PAH-targeted therapies were started.
Haemodynamic response to first-line single- or combination therapy
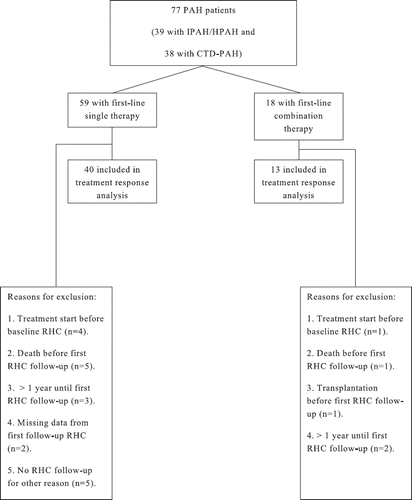
Haemodynamic effects to first-line single- (n = 40) or first-line combination therapy (n = 13), respectively, were investigated and compared in all IPAH/HPAH- and CTD-PAH patients with available data from a RHC before treatment start and at a first RHC follow-up performed within a year from treatment start. The selection of patients for the treatment response analysis is shown in .
Figure 1. Selection of patients for the treatment response analysis. PAH, pulmonary arterial hypertension; IPAH, idiopathic pulmonary arterial hypertension; HPAH, hereditary pulmonary arterial hypertension; CTD-PAH, connective tissue disease-associated pulmonary arterial hypertension; RHC, right heart catheterization.
Prognostic markers
The relationship between haemodynamic- and functional parameters at baseline and outcome, defined as death or transplantation, were investigated in all patients who had gone through RHC at our centre before starting PAH-targeted therapies at time of PAH diagnosis. Data from RHCs before treatment start were available for 71 patients (36 IPAH/HPAH patients and 35 CTD-PAH patients). For one IPAH/HPAH patient there were missing data from the RHC before treatment start. This patient still contributed with data on WHO-FC before treatment start. The relationship between follow-up parameters as well as between differences from baseline to follow-up and outcome were also investigated in patients having gone through follow-up RHC within a year. Such follow-up RHCs were available for 53 patients (29 IPAH/HPAH patients and 24 CTD-PAH patients). For variables associated with outcome, survival rates were compared for patients with infra-median values and patients with values equal to or larger than the median.
Statistics
Survival rates were estimated using the Kaplan–Meier method and differences between groups were investigated using the Log-Rank test. COX regression was used to calculate age-adjusted hazard ratios (HR) for standardized values of potential prognostic markers. Differences in haemodynamic variables before and after treatment start were compared using a paired t-test or Wilcoxon Signed Rank Test. When comparing two groups, a t-test or Rank Sum test was performed. Values are mean ± SD for continuous variables and absolute for categorical variables. P < 0.05 was considered statistically significant. Statistical analyses were performed in SigmaPlot 11 (Systat Software Inc, San Jose, CA, USA) and SPSS (IBM SPSS Statistics 20, IBM Corp., Armonk, NY, USA).
Results
Characteristics of the entire study population
Characteristics at time of first RHC are shown in . 84% of patients with CTD-PAH were diagnosed with systemic sclerosis.
Table I. Characteristics at first right-heart catheterization for the entire study population.
Survival
1-, 2- and 3-year survival rates were 87%, 67% and 54%, respectively, for the entire study population; 89%, 78% and 71%, respectively, for IPAH/HPAH patients; and 84%, 55% and 40%, respectively, for CTD-PAH patients. Survival was better (p = 0.003) for IPAH/HPAH- than CTD-PAH patients ().
Figure 2. 1-, 2- and 3-year survival rates, respectively, for (A) the entire study population, (B) IPAH/HPAH- and CTD-PAH patients, respectively, and the proportion of patients that started first-line single therapy and who were still alive on single therapy at 1-, 2- and 3-years, respectively, from treatment start for (C) the entire study population and (D) IPAH/HPAH- and CTD-PAH patients, respectively. IPAH, idiopathic pulmonary arterial hypertension; HPAH, hereditary pulmonary arterial hypertension; CTD-PAH, connective tissue disease-associated pulmonary arterial hypertension. Our findings support that survival has improved, as compared with untreated patients in the 1991 NIH registry study, which reported a median survival of 2.8 years for patients with primary pulmonary hypertension, and only approximately one year for patients with associated Raynaud phenomenon (A, B). Our data also show that a large proportion of patients who started on first-line single therapy required early treatment escalation (C, D).
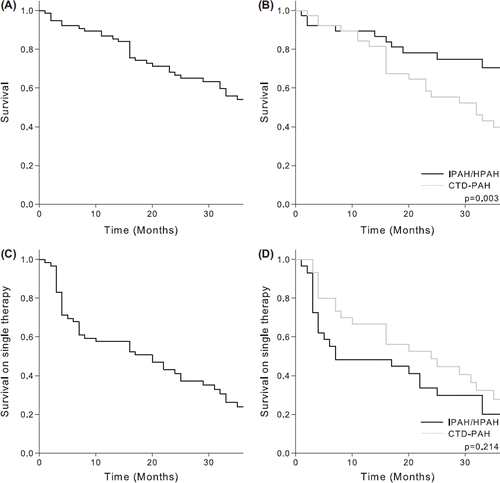
Survival rates were 88%, 65% and 51%, respectively, for patients receiving first-line single therapy; and 82%, 75% and 67%, respectively, for patients receiving first-line combination therapy. Survival rates did not differ (p = 0.712) between the two groups.
Treatment escalation and lung transplantation
The proportion of patients that started on first-line single therapy and that were still alive on single therapy after 1-, 2-, and 3 years were 58%, 41% and 24% for the entire study population; 48%, 34% and 20%, respectively, for IPAH/HPAH patients; and 67%, 49% and 28%, respectively, for CTD-PAH patients. There was no difference (p = 0.214) between IPAH/HPAH- and CTD-PAH patients ().
Already at first RHC follow-up, 37.5% of patients (48% of IPAH/HPAH patients, 26% of CTD-PAH patients) who received first-line single therapy were escalated to combination therapy. Patients who received escalated therapy presented at first RHC follow-up with higher MPAP (p = 0.016), PVRI (p = 0.003), TPG (p = 0.009) and DPG (p = 0.004), and lower CI (p = 0.013), than those who did not receive escalated therapy. There was no significant difference in haemodynamics at baseline between patients who were or were not escalated to combination therapy at first RHC follow-up.
Eight patients were lung transplanted. At the end of the study, seven of them were alive with a mean follow-up time of 77 ± 34 months post-transplantation. One patient died approximately one month post-transplantation.
Haemodynamic response to first-line single- or combination therapy
Mean time to first RHC follow-up were 168 ± 68 and 149 ± 90 days, respectively, for patients who received first-line single- (n = 40) or combination therapy (n = 13). Patients in the first-line single therapy group were treated with bosentan (n = 30), ambrisentan (n = 2), sildenafil (n = 5), iloprost (n = 2) or a CCB (n = 1). Patients in the first-line combination therapy group were treated with bosentan and sildenafil (n = 9), bosentan and iloprost (n = 1), bosentan and epoprostenol (n = 1), bosentan, sildenafil and iloprost (n = 1) or bosentan, sildenafil and treprostinil (n = 1). At the first RHC follow-up, the total daily mean dose of bosentan was 233 mg (27 patients on 125 mg b.i.d., one patient on 62.5 mg b.i.d. and two patients on 31.25 mg b.i.d.) in the first-line single therapy group and 240 mg (12 patients on 125 mg b.i.d. and one patient on 62.5 mg b.i.d.) in the first-line combination therapy group. The total daily mean dose of sildenafil was 60 mg (five patients on 20 mg t.i.d.) in the first-line single therapy group and 82 mg (10 patients on 20 mg t.i.d. and one patient on 100 mg t.i.d.) in the first-line combination therapy group. The total daily mean doses of bosentan and sildenafil did not significantly differ between the first-line single- and first-line combination therapy groups, respectively (p = 0.793 and 0.590, respectively).
Characteristics at baseline and the haemodynamic response to first-line single- or combination therapy, respectively, are shown in . First-line single therapy decreased MPAP (p = 0.048), PVRI (p = 0.04), TPG (p = 0.011) and DPG (p = 0.004). First-line combination therapy decreased MPAP (p = 0.037), PVRI (p = 0.001), TPG (p = 0.002) and DPG (p = 0.004), and increased CI (p = 0.042). First-line combination therapy more greatly decreased PVRI (p = 0.017) than first-line single therapy.
Table II. Baseline characteristics and haemodynamic response to first-line single- and combination therapy, respectively, in IPAH/HPAH and CTD-PAH.
Prognostic markers
At baseline, higher MRAP (p = 0.018), MRAP/CI (p = 0.021) and WHO-FC (p < 0.001) as well as lower 6MWD (p = 0.001) were associated with worse outcome, which was in the present study defined as death or transplantation. At first RHC follow-up within a year, higher PVRI (p = 0.008), as well as lower 6MWD (p = 0.004) were associated with worse outcome. The change from baseline to first RHC follow-up was not associated with outcome for any parameter in the COX regression ().
Table III. Age-adjusted HR/SD for death or transplantation for variables at baseline and first RHC follow-up as well as of changes in variables from baseline to follow-up.
Patients in WHO-FC III-IV at baseline (p = 0.009) and patients with a 6MWD less than 250 m at baseline (p = 0.002) had worse survival rates. Median MRAP (p = 0.179) or MRAP/CI (p = 0.372) at baseline did not separate patients with better or worse survival rates. At first RHC follow-up, patients with a 6MWD less than 325 m (p = 0.026) had worse survival rates. Median PVRI at follow-up did not discriminate (p = 0.565) patients with better or worse survival rates.
Discussion
In accordance with previous large registry studies (Citation5,Citation6) and a meta-analysis (Citation9), we confirmed in our patient cohort improved survival rates in IPAH/HPAH and CTD-PAH since introduction of PAH-targeted therapies, as compared with untreated historic controls (Citation8). Survival rates for newly diagnosed IPAH/HPAH patients were in the present study furthermore even better than those reported for an incident cohort of patients with IPAH-, HPAH- and anorexigen-induced PAH from the French PAH registry (Citation6). Survival for the entire cohort of CTD-PAH patients in the present study furthermore resemble that of newly diagnosed SSc-PAH patients at our centre, recently described by Hesselstrand et al. (Citation7).
Among patients who started on single therapy, the proportions still alive and remaining on single therapy were only 58%, 41%, and 24% at follow-up 1-, 2- and 3-years, respectively, from treatment start. Furthermore, as many as 37.5% of patients who started on single therapy were escalated to combination therapy, already at first RHC follow-up. In addition, approximately as much as 10% of patients were at some point during the follow-up period lung transplanted, underlining the poor prognosis despite of PAH-targeted medical treatments.
At present, the European Society of Cardiology's guidelines recommend first-line single therapy with further addition of drugs at follow-up if prognostic markers are unstable or dissatisfactory (Citation3). Because prognosis is still poor in PAH and a large proportion of patients require early treatment escalation, we retrospectively investigated and compared the response to first-line single- or first-line combination therapy in IPAH/HPAH- and CTD-PAH patients at our centre. There are at present only sparse evidence for the benefit of first-line combination therapy. We found that both patients who received first-line single- and first-line combination therapy improved their haemodynamic status from baseline to first RHC follow-up, although only patients on first-line combination therapy significantly improved CI. Even though single therapy has been shown to improve cardiac function in some randomized controlled trials (Citation18–20), the present study demonstrates that in an unselected, real-life population single therapy is not always enough to immediately improve the cardiac function. Furthermore, patients who received first-line combination therapy more greatly improved PVRI than those who received first-line single therapy. The greater decrease in PVRI and improvement of CI with combination therapy indicates that combination therapy may more potently than single therapy reduce right ventricular afterload and thereby allow for the right ventricle to improve its function. PVRI was furthermore in our PAH cohort interestingly identified as a prognostic marker at RHC follow-up. First-line combination therapy may hence be a way not only to more rapidly and potently improve patients haemodynamically, but also their prognosis.
Only one randomized controlled trial, the BREATHE-2 study, has compared first-line single- with first-line combination therapy. It was found that patients on first-line therapy with iv epoprostenol and bosentan, over a 16-week follow-up period, tended to more greatly improve the measured haemodynamic parameters than patients on first-line therapy with only iv epoprostenol (Citation15). A recent, small observational study by Kemp et al. (Citation16), moreover, found that patients on first-line combination therapy with epoprostenol and bosentan manifested greater improvement in pulmonary vascular resistance, as compared to a matched cohort on single therapy with epoprostenol. In addition, Kemp et al. (Citation16) found a trend for greater survival (p = 0.07) in the combination therapy group, which could, however, not be verified for patients who received first-line combination therapy in the present study. This could reflect the small number of patients in the present study and that our patients who received first-line combination therapy had more severe disease.
In addition, in contrast to the previously published studies, the majority (69%) of patients in the first-line combination therapy group of the treatment response analysis were in the present study treated with a combination of sildenafil and bosentan. In this context, it was recently reported in a press release from the COMPASS-2 trial (ClinicalTrials.gov identifier: NCT00303459) (Citation21), comparing the addition of bosentan or placebo to background sildenafil treatment, that bosentan on top of sildenafil did not meet the primary end-point. This may potentially be explained by bosentan significantly reducing the sildenafil Cmax by 55% and AUC by 63% and that the concomitant sildenafil-induced increase in bosentan Cmax and AUC does not sufficiently compensate (Citation22). Future clinical trials may have to address what combination of PAH-targeted treatments is the most efficient. In summary, the BREATHE-2 study and the study by Kemp et al., as well as our study, indicate beneficial haemodynamic effects of first-line combination therapy in PAH. Prognostic benefits with first-line combination therapy are moreover implied. However, event-driven randomized controlled trials are warranted to clarify whether greater haemodynamic improvement with first-line combination therapy translates into improved outcome and which combination of PAH-targeted treatments is the most efficient. With the introduction of the soluble guanylate cyclase stimulator riociguat (Citation11) and the ERA macitentan (Citation10) for treatment of PAH, studies investigating different combinations of PAH-targeted therapies will be of even greater importance. At present, results are awaited from the AMBITION trial (ClinicalTrials.gov identifier: NCT01178073) (Citation23), comparing first-line single therapy with either the ERA ambrisentan or the PDE5i tadalafil with first-line combination therapy with ambrisentan and tadalafil.
The present study furthermore investigated the association of some selected haemodynamic and functional parameters, at baseline and at first RHC follow-up within a year, with outcome, as defined as death or transplantation. The absolute value of 6MWD was in the present study, and in accordance with previous findings (Citation14,Citation24–28), identified as a strong prognostic marker both at baseline and at first RHC follow-up. The 6MWD was in fact in our study the most powerful prognostic marker also after adjusting for age. A 6MWD less than 250 m at baseline and a 6MWD less than 325 m at follow-up was furthermore associated with worse survival rates. In accordance with recently published meta-analyses of randomized controlled trials in PAH (Citation29–31), the change in our study of 6MWD from baseline to follow-up after initiation of PAH-targeted treatments did not predict outcome. This is of importance as the change in 6MWD has been used as an end-point, often primary end-point, in PAH trials. Results from the SERAPHIN study, the first PAH trial with a morbidity-/mortality end-point, is also in accordance with ours, since the change in 6MWD from baseline to the six-month follow-up was not associated with outcome (Citation32). This underlines the value of morbidity-/mortality end-points in future clinical trials. The lack of a relationship between the change in 6MWD and outcome may reflect that changes in 6MWD induced by PAH-targeted treatments may remain above or below a threshold for increased or reduced risk for poor outcome. WHO-FC was furthermore found to be a strong prognostic marker at baseline, and patients in WHO-FC III-IV at diagnosis had worse survival rates. This verifies the approach of the present European Society of Cardiology guidelines that recommend more aggressive treatment for patients in high functional classes (Citation3). In addition, higher MRAP at baseline was, as in previous studies (Citation8,Citation14,Citation24–28,Citation33,Citation34), associated with worse outcome. In the present study, we furthermore introduce MRAP/CI, a marker of right ventricular function, which increases with rising MRAP and/or falling CI, as a prognostic marker at baseline. As a prognostic marker, MRAP/CI was in our study found to be equivalent to MRAP with a HR/SD of 1.5. CI at baseline was, however, in the present study, and some previous studies (Citation27,Citation28,Citation34), not a significant predictor of outcome. Because MRAP/CI takes into account two parameters of right ventricular function it would be of value to in larger studies investigate whether MRAP/CI is more sensitive than MRAP alone for predicting prognosis in PAH. A high PVRI only tended (95% CI 1.0–2.0, p = 0.062) to be associated with worse outcome at baseline, whereas it was a found to be a significant marker of worse outcome at first RHC follow-up within a year. In addition, MRAP/CI (95% CI 1.0–2.1, p = 0.067) and RVSWI (95% CI 0.4–1.0, p = 0.068) tended to be significant predictors of outcome at first RHC follow-up in the present study. Further larger studies are warranted to investigate their potential as prognostic markers in PAH.
It should be noted that the present study is a small, retrospective, single-centre study. Lack of statistical power may consequently have affected the results. Follow-up data may furthermore be biased due to loss of more severely ill patients who died or were lung transplanted before first RHC follow-up. Population size did not allow for sub-group analysis of treatment response or prognostic markers in IPAH/HPAH and CTD-PAH patients, respectively. Some data points were missing and as this was a retrospective, ‘real-life’ study, patient groups in the treatment response analysis were not matched. Patients were included from 1 January 2000 to 31 December 2010, and available PAH-targeted treatments, as well as the scientific base for treatment decisions, have varied over this period of time. For instance, with respect to the treatment response analysis, it must be noted that a majority (77%) of the patients on first-line combination therapy in the treatment response analysis were diagnosed after 2005, whereas only half of the patients on first-line single therapy were diagnosed after 2005. There were, however, no significant difference in the total daily dose of ERA and PDE5i in the first-line single- and first-line combination therapy groups, respectively, and the greater haemodynamic effects observed with first-line combination therapy was hence not due to higher drug doses. Both statins and sildenafil are substrates for CYP3A4 and statin treatment could therefore theoretically, as previously suggested (Citation35), increase the sildenafil levels. A high proportion of patients concomitantly treated with a statin and sildenafil in the first-line combination therapy group of the treatment response analysis could therefore have biased the results in favour of first-line combination therapy. However, only two patients on sildenafil treatment in the first-line combination therapy group were treated with a statin and it is therefore unlikely that this would have interfered with the results. Nevertheless, the present study illustrates survival, frequency of treatment escalation, treatment response and prognostic markers in an unselected ‘real-life’ population of IPAH/HPAH- and CTD-PAH patients at a single PAH centre.
In conclusion, the present study confirmed that survival has improved for patients with PAH since the introduction of PAH-targeted therapies, but it is nonetheless still poor. We furthermore show that single therapy is often not sufficient for treatment and that PAH patients frequently require early treatment escalation. This warrants for new treatments and treatment regimes. Although it has to be verified in clinical trials, our data indicate that first-line combination therapy, as compared with first-line single therapy, may improve, to a greater extent, haemodynamics from baseline to first follow-up and thereby possibly also survival. Finally, MRAP/CI may be a new additional haemodynamic marker for prognostication in patients with PAH. Additional studies with larger patient cohorts should be encouraged.
Acknowledgements
We acknowledge the support of the staff at the Clinic for Heart Failure and Valvular Disease and the Rheumatology Clinic, Skåne University Hospital, Lund, Sweden.
We acknowledge the financial support of the “ALF”- and Skåne University Hospital Foundations. The foundations have no role in the data collection, analysis or interpretation and have no right in disapproving of the manuscript.
Declaration of interest: Dr. Kylhammar reports unrestricted research grants from The Swedish Society of Pulmonary Hypertension, Actelion Pharmaceuticals Sweden and Pfizer outside the submitted work. Dr. Kylhammar reports personal lecture fees from Actelion Pharmaceuticals Sweden outside the submitted work.
Mrs. Persson reports no relevant conflict of interest.
Dr. Hesselstrand reports no relevant conflict of interest.
Dr. Rådegran reports unrestricted research grants from the “ALF”- and Skåne University Hospital Foundations to complete the work with the present study. Dr. Rådegran reports unrestricted research grants from the Anna-Lisa and Sven-Erik Lundgren- and Maggie Stephens Foundations, as well as Actelion Pharmaceuticals Sweden outside the submitted work. Dr. Rådegran reports personal lecture fees from Actelion Pharmaceuticals Sweden and Sandoz/Novartis outside the submitted work. Dr. Rådegran is and has been primary- or co-investigator in clinical PAH trials for GlaxoSmithKline, Actelion Pharmaceuticals Sweden, Pfizer, Bayer and United Therapeutics, and in clinical post-heart transplantation immunosuppression trials for Novartis. The authors alone are responsible for the content and writing of the paper.
References
- Pietra GG, Capron F, Stewart S, Leone O, Humbert M, Robbins IM, et al. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol. 2004;43: 25S–32.
- Rubin LJ. Primary pulmonary hypertension. N Engl J Med. 1997;336:111–7.
- Galiè N, Hoeper MM, Humbert M, Torbicki A, Vachiéry J-L, Barbera JA, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2009;34: 1219–63.
- Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54: S43–54.
- Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL registry. Chest. 2012;142:448–56.
- Humbert M, Sitbon O, Yaïci A, Montani D, O’Callaghan DS, Jaïs X, et al. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010;36:549–55.
- Hesselstrand R, Wildt M, Ekmehag B, Wuttge DM, Scheja A. Survival in patients with pulmonary arterial hypertension associated with systemic sclerosis from a Swedish single centre: prognosis still poor and prediction difficult. Scand J Rheumatol. 2011;40:127–32.
- D’Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115:343–9.
- Galiè N, Manes A, Negro L, Palazzini M, Bacchi-Reggiani ML, Branzi A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J. 2009;30:394–403.
- Pulido T, Adzerikho I, Channick RN, Delcroix M, Galiè N, Ghofrani HA, et al. Macitentan and Morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809–18.
- Ghofrani HA, Galiè N, Grimminger F, Grünig E, Humbert M, Jing Z-C, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369:330–40.
- Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327:76–81.
- Sitbon O, Humbert M, Jaïs X, Ioos V, Hamid AM, Provencher S, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111:3105–11.
- Nickel N, Golpon H, Greer M, Knudsen L, Olsson K, Westerkamp V, et al. The prognostic impact of follow-up assessments in patients with idiopathic pulmonary arterial hypertension. Eur Respir J. 2012;39:589–96.
- Humbert M, Barst RJ, Robbins IM, Channick RN, Galiè N, Boonstra A, et al. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Respir J. 2004;24:353–9.
- Kemp K, Savale L, O’Callaghan DS, Jaïs X, Montani D, Humbert M, et al. Usefulness of first-line combination therapy with epoprostenol and bosentan in pulmonary arterial hypertension: an observational study. J Heart Lung Transplant. 2012;31:150–8.
- ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med. 2002;166:111–7.
- Channick RN, Simonneau G, Sitbon O, Robbins IM, Frost A, Tapson VF, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet. 2001;358:1119–23.
- Galiè N, Rubin LJ, Hoeper MM, Jansa P, Al-Hiti H, Meyer GMB, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY-study): a double-blind, randomised controlled trial. Lancet. 2008;371:2093–100.
- Galiè N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch DB, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148–57.
- Actelion. Actelion provides an update on the bosentan study - COMPASS-2 [Internet]. Available from: http://www.actelion.com/en/our-company/news-and-events/index.page?newsId = 1769001 [updated March 17 2014; cited 2014 April 17].
- Venitz J, Zack J, Gillies H, Allard M, Regnault J, Dufton C. Clinical pharmacokinetics and drug-drug interactions of endothelin receptor antagonists in pulmonary arterial hypertension. J Clin Pharmacol. 2012;52:1784–805.
- GlaxoSmithKline. AMBITION: A randomized, multicenter study of first-line ambrisentan and tadalafil combination therapy in subjects with pulmonary arterial hypertension (PAH) [Internet]. 2010- [updated April 15 2014]. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01178073.
- Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122:156–63.
- Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial hypertension: insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL). Circulation. 2010;122: 164–72.
- Kane GC, Maradit-Kremers H, Slusser JP, Scott CG, Frantz RP, McGoon MD. Integration of clinical and hemodynamic parameters in the prediction of long-term survival in patients with pulmonary arterial hypertension. Chest. 2011;139: 1285–93.
- Sitbon O, Humbert M, Nunes H, Parent F, Garcia G, Hervé P, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol. 2002;40:780–8.
- Provencher S, Sitbon O, Humbert M, Cabrol S, Jaïs X, Simonneau G. Long-term outcome with first-line bosentan therapy in idiopathic pulmonary arterial hypertension. Eur Heart J. 2006;27:589–95.
- Savarese G, Paolillo S, Costanzo P, D’Amore C, Cecere M, Losco T, et al. Do changes of 6-minute walk distance predict clinical events in patients with pulmonary arterial hypertension? A meta-analysis of 22 randomized trials. J Am Coll Cardiol. 2012;60:1192–201.
- Gabler NB, French B, Strom BL, Palevsky HI, Taichman DB, Kawut SM, et al. Validation of 6-minute walk distance as a surrogate end point in pulmonary arterial hypertension trials. Circulation. 2012;126:349–56.
- Fritz JS, Blair C, Oudiz RJ, Dufton C, Olschewski H, Despain D, et al. Baseline and follow-up 6-min walk distance and brain natriuretic peptide predict 2-year mortality in pulmonary arterial hypertension. Chest. 2013;143:315–23.
- Galiè N, Channick RN, Delcroix M, Ghofrani HA, Jansa P, Le Brun F-O, et al. Sustained effect of macitentan, a novel oral endothelin receptor antagonist, on exercise capacity and the association of its measure with long-term outcomes in pulmonary arterial hypertension. Eur Heart J. 2013;34(Abstract supplement):186.
- Thenappan T, Shah SJ, Rich S, Tian L, Archer SL, Gomberg-Maitland M. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J. 2010;35:1079–87.
- McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation. 2002;106:1477–82.
- Wilkins MR, Ali O, Bradlow W, Wharton J, Taegtmeyer A, Rhodes CJ, et al. Simvastatin as a treatment for pulmonary hypertension trial. Am J Respir Crit Care Med. 2010;181:1106–13.