
Abstract
Objectives. It has previously been demonstrated that 15-min continuous insulin infusion at immediate reperfusion affords cardioprotection. This study sought to reduce the treatment time of insulin and test if intermittent insulin infusions can mimic ischemic postconditioning. Design. In a Langendorff perfused rat heart model of regional ischemia, hearts were at the onset of reperfusion subjected to either 5- or 1-min continuous insulin infusion or 3 × 30 s intermittent insulin infusions (InsPost); with or without inhibitors of Akt (SH-6), p70s6-kinase (rapamycin), mitochondrial ATP-sensitive potassium channels (5-hydroxydecanoic acid [5-HD]), or a scavenger of reactive oxygen species (ROS; 2-mercaptopropionyl glycine [MPG]). Infarct size is expressed as percent of area at risk and presented as mean ± standard error of the mean or s.e.m. Results. Only InsPost was able to reduce infarct size compared with controls (InsPost 33 ± 6% vs. Ctr 52 ± 4%, p < 0.05.). This cardioprotection was abrogated by co-administering SH-6, rapamycin, 5-HD, or MPG. (InsPost + SH-6 56 ± 9%, InsPost + Rapa 55 ± 8%, InsPost + 5-HD 56 ± 7%, InsPost + MPG 60 ± 3% vs. InsPost 33 ± 6% p < 0.05). These results were corroborated by a significant increase in phosphorylated Akt and p70s6k in the InsPost group compared with controls. Conclusion. Short intermittent insulin infusions can mimic ischemic postconditioning and reduce myocardial infarct size via Akt/p70s6k and mKATP channels/ROS-dependent signaling.
Introduction
It has previously been demonstrated that insulin can afford cardioprotection from ischemia–reperfusion-induced injury when administered at the immediate onset of reperfusion (Citation1–3). Recent evidence also demonstrates that insulin can alleviate chronic adverse changes in postischemic cardiac structure and function (Citation4). The binding of insulin to its receptor activates a series of downstream prosurvival kinases, collectively called the Reperfusion Injury Salvaging Kinases (RISK) pathway, which plays a crucial role in both ischemic preconditioning (IPC) and ischemic postconditioning (IPost) (Citation5,Citation6), whereby short alternating cycles of ischemia and reperfusion are applied immediately before or after a prolonged ischemic event, respectively. Activation of phosphoinositide 3-kinase (PI3K) and Akt is central in RISK signaling, which when in a phosphorylated state activates several downstream targets such as p70 ribosomal S6 protein kinase (p70s6k) (Citation3), endothelial NO synthase (eNOS) (Citation7), and glycogen synthase kinase 3 β or GSK3β (Citation8). The end result of RISK activation is preventing or delaying opening of mitochondrial permeability transition pores or mPTPs during early reperfusion, and thus, preventing the associated cell death (Citation9).
Results also indicate that insulin only needs to be administered for the first 15 min of reperfusion to achieve cardioprotection, and if the treatment is delayed 15 min the protection is lost. The same study also indicates that Akt is significantly phosphorylated after only 2 min of insulin administration at reperfusion (Citation2). As Akt phosphorylation plays a critical role in conveying insulin cardioprotection, it should, theoretically, be possible to reduce the treatment time from 15 min. This study therefore explores the cardioprotective potential of shorter insulin treatment protocols. Secondly, we explored a strategy of intermittent insulin infusions in an attempt to shorten treatment time and lower treatment dose by mimicking the treatment protocol of ischemic postconditioning. Both we and others have previously been successful with this conditioning strategy (Citation10,Citation11).
Our final objective was to assess the role of mitochondrial ATP-sensitive potassium (mKATP) channels in insulin treatment at reperfusion. These channels are thought to open at reperfusion in response to IPost (Citation12,Citation13), IPC (Citation14,Citation15), and several other pharmacological treatments (Citation16,Citation17), creating a reactive oxygen species (ROS) burst which phosphorylates and translocates protein kinase Cϵ to the mitochondria, leading to cardioprotection. Previous results from others where insulin was used to precondition hearts or myocytes, however, failed to show any involvement of mKATP channels or the associated mitochondrial ROS burst (Citation18,Citation19), but this has as of yet not been tested at reperfusion.
Methods
Langendorff perfusion procedure
All experiments were approved by the Norwegian State Commission for Laboratory Animals, and carried out in accordance with the European Communities Council Directive of 1986 (2010/63/EEC). Male Wistar rats (300–350 g) fed a standard diet ad libitum were heparinized (200 IU) and anesthetized with sodium pentobarbital (50 mg/kg) i.p. The hearts were excised, placed in ice-cold Krebs–Henseleit buffer (KHB), and rapidly mounted onto a constant-pressure Langendorff perfusion system (80 mmHg). The KHB (pH: 7.4, oxygenated with 95% O2/5% CO2) contained in mM: 118 NaCl, 25.2 NaHCO3, 4.7 KCl, 1.84 CaCl2, 1.21 KH2PO4, 1.22 MgSO4, and 11 glucose. A water-filled latex balloon, connected to a hydrostatic pressure transducer (TrueWave, Edwards Lifesciences, USA) and coupled to a data acquisition system (PowerLab 8/30, Chart Pro software-MLS250), was inserted into the left ventricle (LV) through an incision in the left auricle, and inflated to set a left ventricular end diastolic pressure of 5–10 mmHg during stabilization. The ensuing experiments were performed under isovolumetric conditions, and recordings of LV developed pressure (LVDP) and heart rate were made and used for calculation of rate pressure product (RPP). Coronary flow (CF) was measured by timed collection of effluent over 30 s at each sampling point. Myocardial temperature (37°C) was monitored by inserting a thermo-probe into the pulmonary artery. A 3–0 silk suture was passed around the main branch of the left coronary artery, and the ends were threaded through a small vinyl tube to form a snare. Regional ischemia (RI) was achieved by pulling the snare and confirmed by a substantial fall in both LVDP and CF. Reperfusion was achieved by releasing the snare.
Measurement of ischemic risk zone and infarct size
Infarct size (IS) and area at risk (AAR) measurements were performed according to the gold standard (Citation20). At the end of each experiment, the silk suture was securely tightened and a 0.2% (w/v) suspension of Evans Blue infused to demarcate the risk zone (Duke Scientific Corp., Palo Alto, CA, USA). Thereafter the hearts were frozen (− 20°C) overnight and cut into 2-mm-thick slices from apex to the atrioventricular groove, thawed and stained with 1% triphenyltetrazolium chloride (TTC) in phosphate buffer (pH: 7.4) at 37°C for 15 min, before fixation in 4% formalin to enhance the contrast of the stain. The area of the LV, the infarcted area (IS, TTC negative), and the risk zone (AAR, blue) were determined using a computerized planimetry program (Planimetry+ v2.0; Erik Traasdahl, ENK, Norway) and multiplied by slice thickness (2 mm) to give calculated volumes. IS is expressed as the volume of IS/AAR(%) and risk zone is expressed as the volume of AAR/LV(%) ().
Table I. Ratio of volume of AAR and LV by Evans Blue staining.
Experimental protocol
All hearts were stabilized for 20 min before being subjected to 30-min RI and 120-min reperfusion (). Baseline values for functional parameters were obtained after 18 min of stabilization. Initially, the temporal effects of insulin administration [0.3 mU ml-1 (Citation2)] (Novo Nordisk A/S, Bagsværd, Denmark) were studied by administering insulin for 15 min (InsR + 15), 5 min (InsR + 5), or 1 min (InsR + 1) at the start of reperfusion. Subsequently, the ability of insulin to mimic ischemic postconditioning was studied by administering insulin [0.3 mU ml-1] for 3 × 30 s at immediate reperfusion (). Furthermore, to explore if the pro-survival RISK signaling pathway was underlying the cardioprotective effect of insulin treatment, the following inhibitors ± insulin treatment were administered for 15 min at reperfusion (): Akt inhibitor [SH-6; 10 μM (Citation11)] and the mTOR kinase inhibitor rapamycin [Rapa; 1 nM (Citation2)] (both from Calbiochem). In order to investigate the involvement of mKATP channels and ROS in insulin-induced signaling at reperfusion, the putatively selective inhibitor of mKATP channels, 5-hydroxydecanoic acid [5-HD; 100 μM (Citation14)] and the free radical scavenger, 2-mercaptopropionyl glycine [MPG; 1 mM (Citation14)] was applied for 15 min at reperfusion with or without InsPost (both from Sigma-Aldrich).
Figure 1. Experimental protocol. Stab = stabilization; RI = regional ischemia; Open bars = KHB buffer perfusion; InsR + 1 = insulin [0.3 mU ml-1] for the first 1 min of reperfusion; InsR + 5 = insulin for the first 5 min of reperfusion; InsR + 15 = insulin for the first 15 min of reperfusion; InsPost = insulin postconditioning, alternating cycles of 3 × 30 s with insulin and KHB infusion; IPost = ischemic postconditioning, 3 × 30 s of global ischemia; The inhibitors (SH-6, Rapa, MPG, and 5-HD) were co-administered with or without InsPost.
![Figure 1. Experimental protocol. Stab = stabilization; RI = regional ischemia; Open bars = KHB buffer perfusion; InsR + 1 = insulin [0.3 mU ml-1] for the first 1 min of reperfusion; InsR + 5 = insulin for the first 5 min of reperfusion; InsR + 15 = insulin for the first 15 min of reperfusion; InsPost = insulin postconditioning, alternating cycles of 3 × 30 s with insulin and KHB infusion; IPost = ischemic postconditioning, 3 × 30 s of global ischemia; The inhibitors (SH-6, Rapa, MPG, and 5-HD) were co-administered with or without InsPost.](/cms/asset/896401ef-135e-47b3-b332-5faa6e221716/icdv_a_1071494_f0001_b.gif)
Immunoblot analysis
Myocardial Akt (Phospho-Akt, Ser473, and total-Akt) and p70s6 kinase phosphorylation (Phospho-p70s6k, Thr421/Ser424 and total p70s6k) in the area at risk was determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (all antibodies from Cell Signaling Technology, USA). A separate set of hearts underwent the protocol as previously described () where tissue was collected at 15 min of reperfusion. Hearts perfused with KHB for 20 min served as baseline controls (CtrB), while ischemia–reperfusion controls are CtrRep. Cardiac ventricular tissue was homogenized in a lysis buffer (10 mM Tris at pH of 7.5, 2 mM ethylenediaminetetraacetic acid or EDTA, 330 mM sucrose, and a complete protease inhibitor cocktail from Roche)(Roche Applied Science, Penzberg, Germany) using a Precellys homogenizer (Montigny-le-Bretonneux, France), and tissue debris was removed by centrifugation at 16000 rcf (10 min). Protein quantification, sample preparation (40 μg/lane), and electrophoresis were performed as previously described (Citation2). Ponceau S staining (Sigma, St. Louis, USA) confirmed successful transfer. Densitometric analysis was performed using Quantity One software (Bio-Rad, CA, USA), and phosphorylation of Akt and p70s6k was expressed as the ratio between phosphorylated and total protein levels. GAPDH expression was determined to ensure equal loading.
Statistical analysis
Values are presented as mean ± standard error of the mean (s.e.m). IS, AAR, and SDS-PAGE results were tested for group differences by one-way analysis of variance (ANOVA) combined with Fisher's post hoc test. Comparisons of LVDP, CF, and RPP both within and between groups were tested with mixed ANOVA combined with Tukey's post hoc test for significant differences. We tested stabilization versus RI to confirm equal degree of ischemia between groups, and tested % recovery of 18-min stabilization at 5, 30, and 120 min of reperfusion to determine any differences after treatment. All statistics were performed in IBM SPSS (version 20.0.0). A value of p < 0.05 was considered statistically significant.
Results
Cardioprotection from insulin reperfusion therapy is temporally dependent
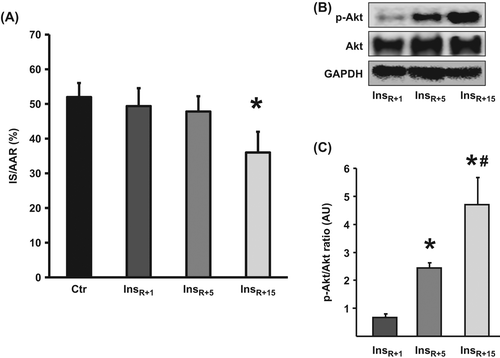
Continuous insulin treatment for the first 5 or 1 min did not reduce IS compared with controls (InsR + 1 49 ± 5%, InsR + 5 48 ± 4% vs. Ctr 52 ± 4%, ns) (), while 15-min treatment did (InsR + 15 36 ± 6% vs. Ctr 52 ± 4%, p < 0.05) (). These findings were corroborated by results from Akt immunoblots where InsR + 15 showed significantly increased levels of phosphorylated Akt compared with InsR + 5 and InsR + 1 (p < 0.05, and C), and InsR + 5 showed twofold higher Akt phosphorylation compared with InsR + 1 (p < 0.05, and ).
Figure 2. The temporal effect of insulin administration for 1, 5, or 15 min at reperfusion. (A) Only InsR + 15 was able to significantly reduce IS versus Ctr. IS is expressed as % of AAR. Bars represent mean ± s.e.m. (n ≥ 6) *p < 0.05 versus Ctr. (B and C) In a parallel set of hearts, Akt phosphorylation was determined at 15-min reperfusion. The longer the duration of insulin therapy, the higher the levels of phosphorylated Akt. Akt phosphorylation at (Ser473) is expressed in arbitrary units (AU) as a ratio relative to total Akt. GAPDH verifies equal loading. Bars represent means ± s.e.m (n ≥ 3).* p < 0.05 versus InsR + 1. #p < 0.05 versus InsR + 5.
Insulin postconditioning is equally cardioprotective as ischemic postconditioning or InsR + 15
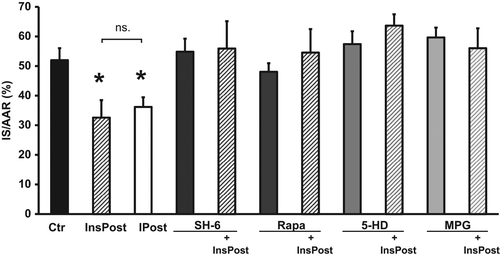
Administration of insulin in bursts of 3 × 30 s at the onset of reperfusion, significantly reduced IS as compared with control hearts (InsPost 33 ± 6% vs. Ctr 52 ± 4%, p < 0.05), but was not statistically different from ischemic postconditioning or InsR + 15 (IPost 36 ± 3%, InsR + 15 36 ± 6% vs. InsPost 33 ± 6%, ns) ( and ). To rule out any possible effects of the conditioning protocol itself, an additional group was administered KHB for 3 × 30 s at reperfusion from a separate reservoir to serve as sham control, and it was not different from Ctr (data not shown).
Figure 3. Insulin postconditioning is equally cardioprotective as ischemic postconditioning and the protection is abrogated by inhibitors of Akt, p70s6k, or mKATP channels, or a ROS scavenger. InsPost showed similar degree of cardioprotection as ischemic postconditioning (IPost). Cardioprotection from InsPost was abrogated by inhibiting Akt (SH-6), mTOR/p70s6k (Rapa), or mKATP channels (5-HD), or by scavenging ROS (MPG). Neither of the inhibitors affected IS alone. IS is expressed as % of AAR. Bars represent mean ± s.e.m. (n ≥ 6) *p < 0.05 versus Ctr.
The infarct sparing effect of insulin postconditioning is abrogated by inhibitors of Akt, p70s6k, or mKATP channels, or ROS scavenging
When inhibitors of Akt, p70s6k, or mKATP channels or a ROS scavenger were co-administered with insulin postconditioning, the cardioprotection was lost (InsPost + SH-6 56 ± 9%, InsPost + Rapa 55 ± 8%, InsPost + 5-HD 64 ± 4%, InsPost + MPG 56 ± 7% vs. InsPost 33 ± 6%, p < 0.05) (). The inhibitors/scavenger had no effect on IS themselves (SH-6 55 ± 4%, Rapa 48 ± 3%, 5-HD 57 ± 4%, MPG 60 ± 3% vs. Ctr 52 ± 4%, ns) ().
Functional parameters are shown in . There were no major group differences at baseline or RI, and all groups had a significant fall in LVDP, CF, and RPP at RI, thus confirming that all groups received sufficient and similar degree of ischemia. The effects of treatment on IS were only marginally reflected in functional data (CF significantly higher in InsR + 15 vs. Ctr at 120 min rep). This is often the case in models of RI, where persistent stunning is thought to obscure the functional effects of more viable tissue in the cardioprotected groups (Citation21).
Table II. Functional parameters recorded during the experimental protocol.
Insulin postconditioning increases Akt and p70s6 phosphorylation during early reperfusion
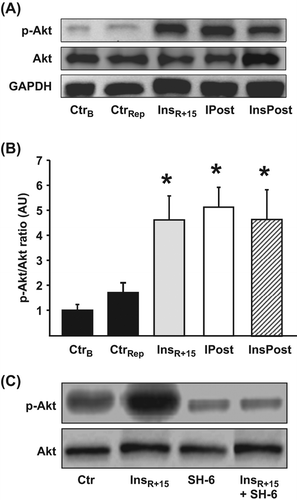
Insulin is known to activate the pro-survival kinase Akt, and we have previously shown that reperfusion therapy with insulin maintains Akt phosphorylation at Ser473 during early reperfusion (Citation2). Insulin postconditioning also maintained Akt in a phosphorylated state as compared with vehicle-treated control hearts at 15-min reperfusion ( and ). The degree of Akt phosphorylation at Ser473 by InsPost was comparable to Akt phosphorylation induced by IPost and InsR + 15 ( and ). The SH-6 inhibitor is a phosphatidylinositol (PI) analog that inhibits Akt without affecting the activity of the upstream kinase PDK-1 (Citation22) and also prevents phosphorylation of Akt at Ser473 (Citation9,Citation23). Administration of SH-6 significantly reduced Akt phosphorylation in hearts exposed to insulin therapy at immediate reperfusion (), validating SH-6 as an Akt inhibitor.
Figure 4. Phosphorylation status of myocardial Akt in hearts exposed to insulin postconditioning. (A) Representative immunoblots of Akt phosphorylation (Ser473) and total Akt, demonstrating the effects of treatment with InsPost compared with ischemic postconditioning (IPost) and 15 min of insulin administration at reperfusion (InsR + 15). CtrB = baseline KHB perfusion for 20 min. CtrRep = ischemia–reperfusion control. GAPDH verifies equal loading. (B) Densitometric analysis of total and phosphorylated Akt immunoblots expressed in AU where p-Akt was expressed as a ratio of total Akt with CtrB = 1. The cardioprotective effect of InsR + 15, IPost, and InsPost was associated with sustained phosphorylation of Akt for the first 15 min of ischemic reperfusion. (C) Blots from insulin-treated hearts confirm that SH-6 indeed abrogates Akt phosphorylation. Bars represent means ± s.e.m (n ≥ 3). *p < 0.05 versus CtrRep and CtrB.
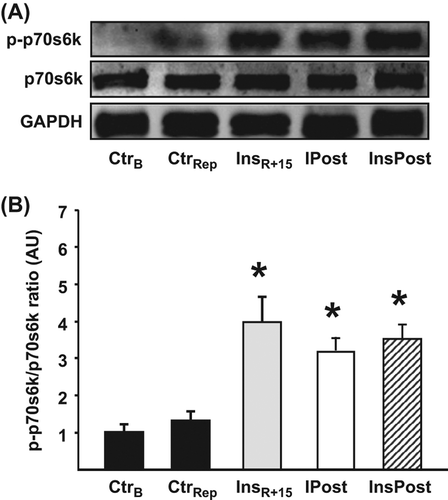
Multiple and divergent pathways activated by Akt are postulated to promote cell survival (Citation24). The mTOR/p70s6k signaling pathway is thought to regulate translational protein synthesis and is central in mammalian cellular growth (Citation25). This signaling pathway also conveys a pro-survival effect (Citation25,Citation26) and is activated by insulin administrated at the time of reperfusion (Citation2,Citation3). In concordance with the elevated levels of phosphorylated Akt, InsPost hearts showed an approximately four-fold increase in p70s6k phosphorylation at Ser421/Thr424 as compared with control hearts, as did IPost and InsR + 15 (p < 0.05, and ).
Figure 5. Phosphorylation status of myocardial p70s6k in hearts exposed to insulin postconditioning. (A) Representative immunoblot of p70s6k phosphorylation (Thr421/Ser424) and total p70s6k demonstrating the effects of InsPost compared with IPost and 15 min of insulin administration at reperfusion (InsR + 15). CtrB = baseline KHB perfusion for 20 min. CtrRep = ischemia–reperfusion control. GAPDH verifies equal loading. (B) Densitometric analysis of total and phosphorylated p70s6k immunoblots expressed in AU were p-p70s6k was expressed as a ratio of total p70s6k with CtrB = 1. The cardioprotective effect of InsR + 15, IPost, and InsPost was associated with sustained phosphorylation of p70s6k for the first 15 min of ischemic reperfusion. Bars represent means ± s.e.m (n ≥ 3). *p < 0.05 versus CtrRep and CtrB.
Discussion
In summary, our data demonstrate that short, continuous insulin treatment for 5 or 1 min at reperfusion fails to offer cardioprotection, while intermittent insulin infusions, given as a mimetic of IPost, afforded cardioprotection to the same extent as InsR + 15 or IPost. Moreover, our data suggest that this cardioprotection is mediated via Akt/p70s6k and mKATP/ROS-dependent RISK survival signaling at reperfusion.
Previously, it has been shown that continuous insulin administration at reperfusion substantially phosphorylated myocardial Akt when the tissue was isolated immediately after 2, 5, or 15 min (Citation2). As Akt seems to be a critical kinase for cardioprotection (reviewed in (Citation27,Citation28)), we therefore expected insulin therapy to be effective in shorter treatment protocols. In the present study, however, all tissues were harvested 15 min into the reperfusion period, and tissue from the shorter insulin treatments showed reduced myocardial Akt phosphorylation (). The blunted Akt phosphorylation could explain the loss of cardioprotection observed in these groups, and taken together with previous reported results, it seems that shorter insulin treatment gives a non-sustained Akt phosphorylation where Akt is phosphorylated acutely, but dephosphorylated upon withdrawal of insulin treatment. How this occurs, however, is yet to be determined.
Recent evidence suggests that co-activation of Akt and AMPK is essential for the cardioprotection afforded by IPC via enhanced myocardial glucose uptake at postischemic reperfusion (Citation29). Ji et al. further show that the reduced ability of IPC to mediate cardioprotection in the streptozotocin-diabetic rat heart could be reversed by insulin, indicating an intrinsic metabolic capacity that could be stimulated by insulin stimulation. Whether pharmacological postconditioning with insulin is dependent on glucose uptake at early reperfusion needs further elucidation. However, previous results have indicated that insulin-mediated Akt-dependent cardioprotection is independent of the presence of glucose at reperfusion, and further, that sarcolemmal glucose transporter type 4 or GLUT4 levels were unchanged in the presence or absence of insulin administration at early reperfusion (Citation2).
In contrast, intermittent infusion of insulin (InsPost), administered as a mimetic of IPost 3 × 30 s, offers equally strong Akt and p70s6k phosphorylation and a similar reduction in IS as InsR + 15, despite a total treatment time of only 1.5 min. It would therefore seem that this cyclic administration of insulin somehow maintains Akt in a phosphorylated state. These findings are in concordance with the study from Penna et al. (Citation10), where a 3-min continuous bradykinin infusion at reperfusion failed to offer cardioprotection, but a 5 × 10 s intermittent infusion succeeded. It is also in concordance with our own findings, where effluents from IPC hearts were cardioprotective when administered for 3 × 30 s at reperfusion (Citation11). In Penna et al.'s study the same pattern could also be mimicked by specific activation of mKATP channels by diazoxide for 5 × 10 s, but not by continuous administration for 3 min.
Controversy exists on whether insulin cardioprotection is mediated via mKATP channels and ROS. LaDisa et al. reported that glucose-insulin-potassium or GIK cardioprotection could be abolished by non-specific inhibition of KATP channels (Citation30), while Baines et al. found no inhibitory effect of either mitochondrial KATP channels or PKCε on insulin preconditioning (Citation18). Juhazhova et al., in an extensive set of experiments, categorized insulin as a “non-sweller,” that is, its protection was independent of mKATP channels, ROS and PKCε translocation, and the associated mitochondrial swelling this would lead to (Citation19). Our results contrast these and indicate that, at least in the setting of reperfusion, intermittent insulin infusions convey cardioprotection via mKATP channels. An important limitation of this interpretation is a series of reports from Hanley et al. showing that 5-HD can be beta-oxidized, and thus create a bottleneck for fatty acid oxidation (Citation31–33), suggesting an alternative explanation for the inhibitory effects of 5-HD. Our preparation, however, used glucose as the only metabolic substrate, so it is uncertain whether this effect pertains to the present study.
The role of ROS in myocardial ischemia–reperfusion damage is well established, and IPost and other cardioprotective treatments have been demonstrated to inhibit ROS generation at reperfusion (Citation13,Citation34,Citation35). However, several studies have reported that pharmacological or mechanical cardioprotective treatments are dependent on ROS signaling (Citation10,Citation12,Citation36,Citation37), creating what Hausenloy et al. coined the free radical signaling paradox (Citation28). Insulin has previously been shown to be cardioprotective by reducing oxidative/nitrative stress (Citation38), and although studies from adipocytes indicate that insulin elicits a burst of hydrogen peroxide that enhances insulin receptor tyrosine phosphorylation, and thus, insulin signaling (Citation39,Citation40), no studies have to date investigated if cardioprotection from insulin treatment at reperfusion is dependent on ROS signaling. In the present study, addition of MPG at reperfusion abrogated the cardioprotection afforded by InsPost, while having no effect on IS alone. This supports a role for ROS signaling in insulin cardioprotection as well, albeit with insufficient evidence for what kind of subspecies, at which concentration and where in the signaling cascade this takes place. However, MPG does not appear to scavenge hydrogen peroxide (Citation41), so this is unlikely to be an effect of hydrogen peroxide on the insulin receptor. A few have reported reduced IS when treating with MPG alone (Citation42–44). The present study, in concordance with several others (Citation10,Citation14,Citation36,Citation45), found no such effect, and this could be related to differences in concentration of MPG or the administration protocol.
There are some important limitations to consider: The Langendorff heart perfusion is isolated from blood, immune system, and neuro-hormonal influence, thus our results could be different in the in vivo setting. We found no correlation between IS and functional recovery at reperfusion. This might be surprising, especially considering the inotropic effect of insulin. However, it is in concordance with others, who have used even higher concentrations of insulin (Citation46), and in general, the lack of correlation between heart function and IS has been observed by several others and is believed to be due to persistent stunning (Citation21,Citation47).
In conclusion, intermittent insulin treatment of 3 × 30 s offers strong cardioprotection in the ex vivo rat heart by activating Akt/p70s6k and mKATP/ROS-dependent RISK survival signaling at reperfusion. In contrast, longer but continuous insulin reperfusion therapy for 5 min offers no cardioprotection.
Acknowledgements
Ingrid Strand is thanked for technical assistance. AKJ is supported by the Bergen Medical Research Foundation, L. Meltzer Foundation, Norwegian Council on Cardiovascular Diseases, and University of Bergen Heart Foundation. LB is supported by the Norwegian Council on Cardiovascular Diseases. EH is supported by Norwegian Research Council.
Declaration of interest: The authors report no conflict of interest. The authors alone are responsible for the content and writing of the paper.
References
- Jonassen AK, Brar BK, Mjos OD, Sack MN, Latchman DS, Yellon DM. Insulin administered at reoxygenation exerts a cardioprotective effect in myocytes by a possible anti-apoptotic mechanism. J Mol Cell Cardiol. 2000;32:757–64.
- Jonassen AK, Sack MN, Mjos OD, Yellon DM. Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circ Res. 2001;89:1191–8.
- Jonassen AK, Mjos OD, Sack MN. p70s6 kinase is a functional target of insulin activated Akt cell-survival signaling. Biochem Biophys Res Commun. 2004;315:160–5.
- Xing W, Yan W, Fu F, Jin Y, Ji L, Liu W, et al. Insulin inhibits myocardial ischemia-induced apoptosis and alleviates chronic adverse changes in post-ischemic cardiac structure and function. Apoptosis. 2009;14:1050–60.
- Hausenloy DJ, Tsang A, Yellon DM. The reperfusion injury salvage kinase pathway: a common target for both ischemic preconditioning and postconditioning. Trends Cardiovasc Med. 2005;15:69–75.
- Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–88.
- Gao F, Gao E, Yue TL, Ohlstein EH, Lopez BL, Christopher TA, Ma XL. Nitric oxide mediates the antiapoptotic effect of insulin in myocardial ischemia-reperfusion: the roles of PI3-kinase, Akt, and endothelial nitric oxide synthase phosphorylation. Circulation. 2002;105:1497–502.
- Vigneron F, Dos Santos P, Lemoine S, Bonnet M, Tariosse L, Couffinhal T, et al. GSK-3beta at the crossroads in the signalling of heart preconditioning: implication of mTOR and Wnt pathways. Cardiovasc Res. 2011;90:49–56.
- Davidson SM, Hausenloy D, Duchen MR, Yellon DM. Signalling via the reperfusion injury signalling kinase (RISK) pathway links closure of the mitochondrial permeability transition pore to cardioprotection. Int J Biochem Cell Biol. 2006;38:414–9.
- Penna C, Mancardi D, Rastaldo R, Losano G, Pagliaro P. Intermittent activation of bradykinin B2 receptors and mitochondrial KATP channels trigger cardiac postconditioning through redox signaling. Cardiovasc Res. 2007;75:168–77.
- Breivik L, Helgeland E, Aarnes EK, Mrdalj J, Jonassen AK. Remote postconditioning by humoral factors in effluent from ischemic preconditioned rat hearts is mediated via PI3K/Akt-dependent cell-survival signaling at reperfusion. Basic Res Cardiol. 2011;106:135–45.
- Penna C, Rastaldo R, Mancardi D, Raimondo S, Cappello S, Gattullo D, et al. Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K + channel and protein kinase C activation. Basic Res Cardiol. 2006;101:180–9.
- Jin C, Wu J, Watanabe M, Okada T, Iesaki T. Mitochondrial K+ channels are involved in ischemic postconditioning in rat hearts. JPS. 2012;62:325–32.
- Hausenloy DJ, Wynne AM, Yellon DM. Ischemic preconditioning targets the reperfusion phase. Basic Res Cardiol. 2007;102:445–52.
- Tsukamoto O, Asanuma H, Kim J, Minamino T, Takashima S, Ogai A, et al. A role of opening of mitochondrial ATP-sensitive potassium channels in the infarct size-limiting effect of ischemic preconditioning via activation of protein kinase C in the canine heart. Biochem Biophys Res Commun. 2005;338:1460–6.
- Perrelli MG, Tullio F, Angotti C, Cerra MC, Angelone T, Tota B, et al. Catestatin reduces myocardial ischaemia/reperfusion injury: involvement of PI3K/Akt, PKCs, mitochondrial KATP channels and ROS signalling. Pflugers Archiv. 2013;465:1031–40.
- Nunez IP, Fantinelli J, Arbelaez LF, Mosca SM. Mitochondrial KATP channels participate in the limitation of infarct size by cariporide. Naunyn-Schmiedeberg's Arch Pharmacol. 2011;383:563–71.
- Baines CP, Wang L, Cohen MV, Downey JM. Myocardial protection by insulin is dependent on phospatidylinositol 3-kinase but not protein kinase C or KATP channels in the isolated rabbit heart. Basic Res Cardiol. 1999;94:188–98.
- Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–49.
- Bell RM, Mocanu MM, Yellon DM. Retrograde heart perfusion: the Langendorff technique of isolated heart perfusion. J Mol Cell Cardiol. 2011;50:940–50.
- Lochner A, Genade S, Moolman JA. Ischemic preconditioning: infarct size is a more reliable endpoint than functional recovery. Basic Res Cardiol. 2003;98:337–46.
- Kozikowski AP, Sun H, Brognard J, Dennis PA. Novel PI analogues selectively block activation of the pro-survival serine/threonine kinase Akt. J Am Chem Soc. 2003;125: 1144–5.
- Stojanovic A, Marjanovic JA, Brovkovych VM, Peng X, Hay N, Skidgel RA, et al. A phosphoinositide 3-kinase-AKT-nitric oxide-cGMP signaling pathway in stimulating platelet secretion and aggregation. J Biol Chem. 2006;281:16333–9.
- Sugden PH, Clerk A. Akt like a woman: gender differences in susceptibility to cardiovascular disease. Circ Res. 2001;88:975–7.
- Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad Sci U S A. 2001;98:9666–70.
- Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–62.
- Mullonkal CJ, Toledo-Pereyra LH. Akt in ischemia and reperfusion. J Invest Surg. 2007;20:195–203.
- Hausenloy DJ, Yellon DM. Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection. Heart Fail Rev. 2007;12:217–34.
- Ji L, Zhang X, Liu W, Huang Q, Yang W, Fu F, et al. AMPK-regulated and Akt-dependent enhancement of glucose uptake is essential in ischemic preconditioning-alleviated reperfusion injury. PloS One. 2013;8:e69910.
- LaDisa JF, Jr., Krolikowski JG, Pagel PS, Warltier DC, Kersten JR. Cardioprotection by glucose-insulin-potassium: dependence on KATP channel opening and blood glucose concentration before ischemia. Am J Physiol Heart Circ Physiol. 2004;287:H601–7.
- Hanley PJ, Mickel M, Loffler M, Brandt U, Daut J. K(ATP) channel-independent targets of diazoxide and 5-hydroxydecanoate in the heart. J Physiol. 2002;542:735–41.
- Hanley PJ, Gopalan KV, Lareau RA, Srivastava DK, von Meltzer M, Daut J. Beta-oxidation of 5-hydroxydecanoate, a putative blocker of mitochondrial ATP-sensitive potassium channels. J Physiol. 2003;547:387–93.
- Hanley PJ, Drose S, Brandt U, Lareau RA, Banerjee AL, Srivastava DK, et al. 5-Hydroxydecanoate is metabolised in mitochondria and creates a rate-limiting bottleneck for beta-oxidation of fatty acids. J Physiol. 2005;562:307–18.
- Kin H, Zhao ZQ, Sun HY, Wang NP, Corvera JS, Halkos ME, et al. Postconditioning attenuates myocardial ischemia-reperfusion injury by inhibiting events in the early minutes of reperfusion. Cardiovasc Res. 2004;62:74–85.
- Sun HY, Wang NP, Kerendi F, Halkos M, Kin H, Guyton RA, et al. Hypoxic postconditioning reduces cardiomyocyte loss by inhibiting ROS generation and intracellular Ca2 + overload. Am J Physiol Heart Circ Physiol. 2005;288:H1900–8.
- Tsutsumi YM, Yokoyama T, Horikawa Y, Roth DM, Patel HH. Reactive oxygen species trigger ischemic and pharmacological postconditioning: in vivo and in vitro characterization. Life Sci. 2007;81:1223–7.
- Downey JM, Cohen MV. A really radical observation–a comment on Penna et al. in Basic Res Cardiol (2006) 101:180–189. Basic Res Cardiol. 2006;101:190–1.
- Ji L, Fu F, Zhang L, Liu W, Cai X, Zheng Q, et al. Insulin attenuates myocardial ischemia/reperfusion injury via reducing oxidative/nitrative stress. Am J Physiol Endocrinol Metab. 2010;298:E871–80.
- Wu X, Zhu L, Zilbering A, Mahadev K, Motoshima H, Yao J, et al. Hyperglycemia potentiates H(2)O(2) production in adipocytes and enhances insulin signal transduction: potential role for oxidative inhibition of thiol-sensitive protein-tyrosine phosphatases. Antioxid Redox Signal. 2005;7:526–37.
- Mahadev K, Motoshima H, Wu X, Ruddy JM, Arnold RS, Cheng G, et al. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol. 2004;24:1844–54.
- Bolli R, Jeroudi MO, Patel BS, Aruoma OI, Halliwell B, Lai EK, et al. Marked reduction of free radical generation and contractile dysfunction by antioxidant therapy begun at the time of reperfusion. Evidence that myocardial “stunning” is a manifestation of reperfusion injury. Circ Res. 1989;65:607–22.
- Horwitz LD, Fennessey PV, Shikes RH, Kong Y. Marked reduction in myocardial infarct size due to prolonged infusion of an antioxidant during reperfusion. Circulation. 1994; 89:1792–801.
- Mitsos SE, Fantone JC, Gallagher KP, Walden KM, Simpson PJ, Abrams GD, et al. Canine myocardial reperfusion injury: protection by a free radical scavenger, N-2-mercaptopropionyl glycine. J Cardiovasc Pharmacol. 1986;8:978–88.
- Mitsos SE, Askew TE, Fantone JC, Kunkel SL, Abrams GD, Schork A, et al. Protective effects of N-2-mercaptopropionyl glycine against myocardial reperfusion injury after neutrophil depletion in the dog: evidence for the role of intracellular-derived free radicals. Circulation. 1986;73:1077–86.
- Tanonaka K, Iwai T, Motegi K, Takeo S. Effects of N- (2-mercaptopropionyl)-glycine on mitochondrial function in ischemic-reperfused heart. Cardiovasc Res. 2003;57: 416–25.
- de Oliveira UO, Bello-Kein A, de Oliveira AR, Kuchaski LC, Machado UF, Irigoyen MC, et al. Insulin alone or with captopril: effects on signaling pathways (AKT and AMPK) and oxidative balance after ischemia-reperfusion in isolated hearts. Fundam Clin Pharmacol. 2012;26:679–89.
- Schulz R, Cohen MV, Behrends M, Downey JM, Heusch G. Signal transduction of ischemic preconditioning. Cardiovasc Res. 2001;52:181–98.