
Abstract
We describe a 74-year-old Japanese man with systemic fibroinflammatory conditions closely resembling those of immunoglobulin G4-related disease (IgG4-RD). Radiology and histology showed characteristics of IgG4-related tubulointerstitial nephritis, despite normal serum IgG4 value and scanty IgG4-positive plasma cell infiltration in each organ. This case suggests that a condition closely mimicking IgG4-RD may develop without IgG4-positive plasma cells and those exceptional cases should also be taken into account in the differential diagnosis of IgG4-RD.
Introduction
Immunoglobulin G4-related disease (IgG4-RD) is a newly recognized fibroinflammatory systemic disease involving multiple organs, for example, pancreas (type 1 autoimmune pancreatitis [type 1 AIP]), biliary tree, salivary glands (SGs), periorbital tissues, kidneys, lungs, lymph nodes (LNs), and aorta [Citation1–3]. Hallmarks of IgG4-RD include serum IgG4 elevation and substantial IgG4-positive plasma cell (PC) infiltration in affected organs [Citation4,Citation5], although around 20% of type 1 AIP patients have normal serum IgG4 levels [Citation6].
IgG4-related kidney disease (IgG4-RKD) usually presents as IgG4-related tubulointerstitial nephritis (TIN) [Citation7–10]. In addition to serum IgG4 elevation and IgG4-postive PC infiltration, IgG4-related TIN has several characteristic morphological and histological features, that is, storiform fibrosis (bird's-eye pattern fibrosis) and regional lesion distribution [Citation7–9]. Although the Japanese Society of Nephrology and Mayo Clinic, respectively, proposed diagnostic criteria for IgG4-RKD based on the above features [Citation11,Citation12], it remains undetermined whether IgG4 molecule per se is always directly implicated in the development of IgG4-related TIN or is only a bystander.
We report a case with a condition closely mimicking IgG4-RD with multiple organ involvement including characteristic IgG4-related TIN, but with normal serum IgG4 value and scanty IgG4-positive PC infiltration. The findings suggest that IgG4 itself is not always responsible for the characteristic picture of IgG4-RD, although this paradox is difficult to explain and will require further investigation and collection of cases.
Case report
A 74-year-old Japanese man was admitted to our hospital for investigation of multiple swollen LNs with hypergammaglobulinemia and eosinophilia. He had been treated for type 2 diabetes mellitus, hypertension, and old cerebral infarction. He had received a left internal carotid artery intimal resection at the age of 64 years and percutaneous coronary interventions at the age of 69 and 71 years. He had allergic rhinitis for 2 years. Seven months before admission, he presented with a 1-week history of dry cough to an affiliated hospital. Laboratory data showed eosinophilia (eosinophil count 1,840/μL) and slightly increased serum IgG levels (1,917 mg/dL; reference, 739–1,649 mg/dL). Computed tomography (CT) of the chest and abdomen revealed mediastinal lymphadenopathy, ground glass opacities and consolidation in the bilateral lungs, and swelling of the pancreatic tail. He was diagnosed with eosinophilic pneumonia from the findings of bronchoalveolar lavage and transbronchial lung biopsy. Bronchial asthma was also diagnosed based on spirometry, and β-2 stimulator inhalation therapy was started. Six weeks before, he had been admitted to the affiliated hospital because of poorly controlled diabetes and eosinophilic pneumonia (eosinophil count 2,751/μL with worsening of pulmonary CT findings) and worsening of generalized lymphadenopathy. Because hematological malignancy was suspected, extensive examinations including bone marrow and submandibular LN biopsies were performed. Neither bone marrow nor submandibular LN biopsies showed any atypical cells or monoclonalities. Positive emission tomography showed accumulation of fluorodeoxyglucose in the parotid and submandibular glands, LNs, lungs, pancreas, kidneys, and prostate (). He was referred to our hospital for further close examination and treatment.
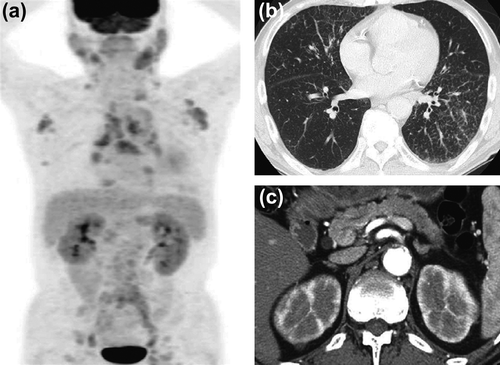
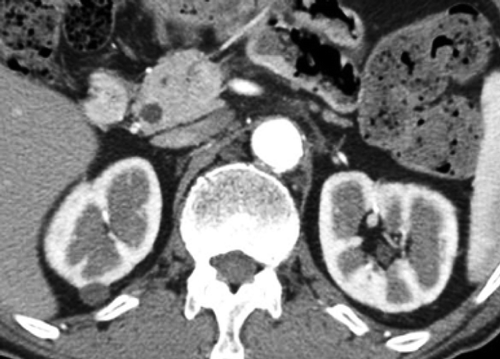
On physical examination, the temperature was 36.2°C, and the blood pressure was 129/89 mmHg. Several bilateral neck, axillary, and inguinal LNs were swollen to 1–2 cm in diameter, respectively. All LNs were soft and non-tender. Lungs were clear to auscultation and abdomen revealed no abnormalities. Laboratory findings included the following: white blood cell count: 5,900/μL with 25% eosinophils (eosinophil count 1,475/μL), hemoglobin: 12.7 g/dL, platelet count: 373,000/μL, aspartate aminotransferase: 35 IU/L, alanine aminotransferase: 38 IU/L, serum creatinine: 0.71 mg/dL, C-reactive protein: 0.2 mg/dL, IgG: 5,593 mg/dL (IgG1 2,700 mg/dL [reference, 320–748 mg/dL], IgG2: 1,020 mg/dL [reference, 208–754 mg/dL], IgG3: 165 mg/dL [reference, 6.6–88.3 mg/dL], IgG4: 10 mg/dL [reference, 4.8–105 mg/dL]), IgE: 352 IU/mL (reference, < 250 IU/mL), C3 40 mg/dL (reference, 44–102 mg/dL), C4 1 mg/dL (reference, 14–49 mg/dL), total hemolytic complement (CH50) 0 U/mL (reference, 31–49 U/mL), rheumatoid factor: 31 IU/mL (reference, < 20 IU/mL), and soluble interleukin-2 receptor (sIL-2R): 2,423 U/mL (reference, 220–530 U/mL). Antinuclear antibodies were slightly positive at a titer of 1:20 with homogeneous pattern. Urinalysis showed negative proteinuria and negative occult blood. The level of urinary N-acetyl-β-D-glucosaminidase was 24.8 U/L (reference, 1–5 U/L) and that of urinary β2-microglobulin was 1,522 μg/L (reference, 16–518 μg/L). Enhanced CT showed submandibular, supraclavicular, axillary, mediastinal, paraaortic, and inguinal LNs swelling, interstitial pneumonia with small nodular lesions and bronchiectasia in the lung, hepatosplenomegaly, patchy hypoattenuated lesions in the pancreas head and body, multiple low-density lesions in the kidneys, and abdominal aortic aneurysm with aortic wall thickening (, ). We performed a renal biopsy, since the radiographic abnormalities were suggestive of IgG4-RKD.
The renal biopsy revealed lymphoplasmacytic infiltration with fibrosis mimicking IgG4-related TIN (). A few infiltrating eosinophils were also noted. In addition, regional lesions with clear borders between affected and unaffected areas with characteristic storiform fibrosis (bird's-eye pattern fibrosis) [Citation7] were revealed. In contrast, the glomeruli and vessels showed only minor abnormalities. Immunofluorescence showed no predominant immunoglobulin and complement deposition, and so we did not perform electron microscopy. Immunostaining revealed dense CD138-positive PC infiltration (). In contrast, IgG4-positive PCs in the most concentrated areas were below 10/hpf, and IgG4-positive/IgG-positive PCs ratio was also below 40% (). IgG1 and IgG2-positive PCs were predominant (data not shown).
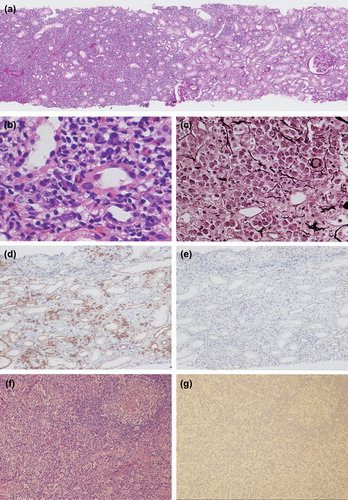
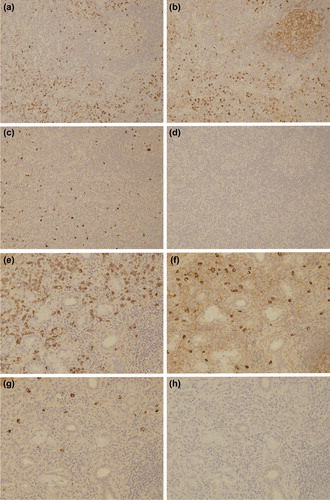
We further performed inguinal LN and small SG biopsies. The inguinal LN biopsy showed decreases of lymphoid follicles, atrophic germinal center with discrete mantle zone, and expansion of the interfollicular area (). In addition, high endothelial venules were prominent and there was polymorphous cellular infiltration; small lymphocytes, PCs, and atypical immunoblasts. Neither light chain restriction nor immunoglobulin heavy-chain rearrangement was detected. These light microscopic features were similar to those of IgG4-related lymphadenopathy (type III: interfollicular expansion and immunoblastosis) [Citation13]. Although complete exclusion of early-stage T cell lymphoma was difficult because TCR-γ gene rearrangement was detected, totally negative immunostainings using diagnostic markers including CXCL-13, PD-1, and CD10 suggested that the diagnosis of angio-immunoblastic T cell lymphoma was very unlikely. The small SG biopsy showed dense lymphoplasmacytic infiltration with moderate fibrosis around ducts. These findings were rather more typical for IgG4-RD than for Sjögren's syndrome. However, the absolute number of infiltrating IgG4-positive PCs was very low, and the results of immunostaining of LN and small SG with IgG subclass were as follows: LN; IgG1 244/hpf, IgG2 131/hpf, IgG3 46/hpf, IgG4 0/hpf; small SG; IgG1 183/hpf, IgG2 62/hpf, IgG3 25/hpf, and IgG4 0/hpf ( and ).
Although the patient did not meet either the comprehensive diagnostic criteria for IgG4-RD or diagnostic criteria for IgG4-RKD [Citation11,Citation12,Citation14], we administered 30 mg/day (0.5 mg/kg/day) of prednisolone (PSL) because the possibility of lymphoma was considered quite low. After the administration of PSL, the swollen LNs shrank rapidly. Two weeks thereafter, enhanced CT demonstrated improvement of the generalized lymphadenopathy and interstitial pneumonia, disappearance of multiple low-density lesions in the kidneys, and reduced size of the pancreas (). Laboratory data were also dramatically improved within 4 weeks as follows: eosinophil count 51/μL, IgG 1,008 mg/dL, IgE 26 IU/mL, C3 55 mg/dL, C4 21 mg/dL, CH50 42 U/mL, and sIL-2R 272 U/mL. The dose of PSL was gradually tapered to 5 mg/day, which was then adopted as the maintenance dose. No flare has been observed for 42 months.
Discussion
Despite the absence of serum IgG4 elevation and IgG4-positive PCs infiltration, the present case showed a systemic fibroinflammatory condition with multiple organ involvement remarkably similar to that of IgG4-RD. It is noteworthy that the kidney examination revealed many of the specific features of IgG4-RKD including multiple hypodense lesions in radiographic examination and lymphoplasmacytic infiltrates with storiform fibrosis (bird's-eye pattern fibrosis), eosinophil infiltration, and regional lesion distribution in histopathological examination [Citation7–9]. Moreover, various negative findings of IgG4-RKD were absent including granuloma, fibrinoid necrosis, neutrophil infiltration, atypical cells, and severe tubulitis which are suggestive of other inflammatory or malignant diseases as well. A favorable response to corticosteroid was also consistent with IgG4-RKD [Citation15]. In spite of these similarities to IgG4-RD, the patient did not meet either the comprehensive diagnostic criteria for IgG4-RD or the proposed diagnostic criteria for IgG4-RKD because serum IgG4 elevation and IgG4-positive PC infiltration are the essential in these diagnostic criteria [Citation11,Citation12,Citation14].
Indeed, elevated serum IgG4 level is a major hallmark of IgG4-RD [Citation5]. However, recent studies have shown that it is not uniformly present in type 1 AIP (sensitivity 68 to 81%) [Citation16,Citation17], and cases of IgG4-RD with normal serum IgG4 levels have also been reported. Kamisawa et al. compared the characteristics of serum IgG4-negative type 1 AIP with those of serum IgG4-positive type 1 AIP, although kidney involvement was not noted [Citation6]. In serum IgG4-negative type 1 AIP patients, morphological, and histological features were somewhat different with segmental swelling of the pancreas, less IgG4-positive PC infiltration, and fewer extrapancreatic lesions [Citation6]. In addition, Zen and Nakanuma showed that the number of involved organs influences serum IgG4 levels [Citation18]. Moreover, in patients with IgG4-related retroperitoneal fibrosis, low serum IgG4 levels have been observed probably because of the presence of advanced disease with dominant fibrosis. These findings suggest that serum IgG4 level depends on the extent and phase of lesions in IgG4-RD. Thus, it is unnatural in our case that serum IgG4 level is within the normal range despite that he had multiple organ involvements and each kidney, LN and small SG involvement was not in an advanced phase with predominant fibrosis but rather in a relatively early phase with PC-rich conditions.
Another important hallmark of IgG4-RD is dense IgG4-positive PC infiltration in affected organs. Hence, an IgG4-positive PC/IgG-positive PC ratio or absolute number of tissue-infiltrating IgG4-positive PCs has been adopted as a critical diagnostic criterion with high sensitivity and high specificity [Citation17,Citation19,Citation20]. To date, only one case report from Makiishi et al. was published recently which showed scant IgG4-positive PC infiltration in the affected organs whose clinical picture was undistinguishable from that of IgG4-RD [Citation21]. Our case was quite similar to Makiishi's case with submandibular gland, kidney, LN, pancreas, and periaortic lesions with marked hypocomplementemia, hypergammaglobulinemia, and normal serum IgG4 level. In our case, not only the kidney but also all other biopsied organs such as LN and small SGs had few IgG4-positive PC infiltrates. Although the similar diagnostic significance of small SG biopsy in comparison with other typically involved organs is not widely accepted yet, it would be very difficult to attribute the scarce IgG4-positive PC infiltrates noted in all biopsied organs in this case to mere sampling error. Because this study and Makiishi's cases were very difficult to diagnose using the already available diagnostic criteria of IgG4-RD, prudent clinical and imaging evaluations are necessary when such cases with clinical features very similar to those of IgG4-RD and without evidence of involvement of IgG4 are encountered.
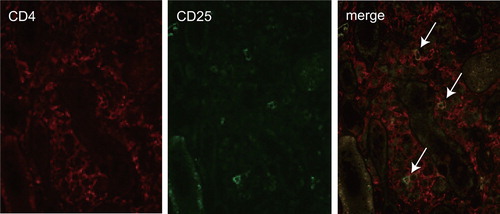
In order to understand what actually makes the characteristic lesions of IgG4-RD, it might be useful to clarify whether the immunological background in our case is similar to that of IgG4-RD in spite of scant IgG4 production. Predominant activation of T helper type 2 (Th2) and regulatory T (Treg) cells is characteristic of IgG4-RD including IgG4-related TIN, type 1 AIP and IgG4-related Mikulicz disease [Citation22–24]. Th2 cells produce interleukin (IL)-4, IL-5, IL-13, and these cytokines induce eosinophilia and production of IgE. In addition, Treg cells produce IL-10 and transforming growth factor-beta (TGF-β). IL-4 and IL-10 induce a class switch to IgG4, and TGF-β induces fibrosclerosis. In the present case, a history of allergies, eosinophilia, serum IgE elevation and eosinophil infiltration point to a Th2-dominant immune response. Furthermore, CD4 and CD25 double-positive cell infiltration in the kidney showed Treg cells involvement (). Certainly, these immunological conditions are insufficient to conclude the diagnosis of IgG4-RD because it remains undetermined whether Treg cell infiltration is useful for the diagnosis of IgG4-RD. However, in our case, these findings suggest that not IgG4 molecule per se but Th2- and Treg-dominant immune response contributes to the formation of IgG4-RKD-like lesions. Although we could not clarify why a class switch to IgG4 was not induced in spite of activation of Th2 and Treg cells, the possibility of some abnormality of IL-10 signals was surmised.
Figure 5. Double immunostaining of CD4 and CD25 in tubulointerstitial lesion. Mononuclear cells positive for CD4 (red) or CD25 (green) are detected. The merge image showed a few cells doubly positive for CD4 and CD25 in an area of interstitial cell infiltration lesion (arrows).
Exclusion of malignant lymphoma was also of major importance in our case. Histopathological, immunohistochemical, and biomolecular analyses of biopsied LN could not completely exclude the possibility of angioimmunoblastic T cell lymphoma or angioimmunoblastic lymphadenopathy with dysproteinemia. However, his very benign clinical course in addition to the presence of several extranodal lesions such as pancreatitis and TIN, all of which would be atypical for lymphoma, rather supports the diagnosis within the IgG4-RD spectrum.
In summary, our case suggests that a clinical picture and characteristic renal histology closely resembling those of IgG4-RKD can be induced even in the absence of IgG4 involvement. We cannot completely exclude the possibility that our patient did not meet the diagnostic criteria for IgG4-RD because of mere sampling error or the late phase of the disease. However, histopathological findings indicating relatively early phase with PC-rich lesions suggest that scanty IgG4-positive PC infiltration of all three biopsied organs is not mere sampling error. Thus, we conclude that a condition closely mimicking IgG4-RD may develop without IgG4-positive PCs. In the meantime until additional information becomes available and more cases are collected, it seems reasonable to treat such cases with steroid administration after careful examination to exclude other pathologies especially malignancy.
Acknowledgments
This work was supported by Intractable Diseases, the Health and Labor Sciences Research Grants from Ministry of Health, Labor and Welfare, Japan. We thank John Gelblum for his critical reading of the manuscript.
Conflicts of interest
None.
References
- Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366(6):539–51.
- Hamed G, Tsushima K, Yasuo M, Kubo K, Yamazaki S, Kawa S, et al. Inflammatory lesions of the lung, submandibular gland, bile duct and prostate in a patient with IgG4-related multifocal systemic fibrosclerosis. Respirology. 2007;12(3):455–7.
- Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T, et al. A novel clinical entity, IgG4-related disease (IgG4RD): general concept and details. Mod Rheumatol. 2012;22(1):1–14.
- Hamano H, Kawa S, Ochi Y, Unno H, Shiba N, Wajiki M, et al. Hydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis. Lancet. 2002;359(9315):1403–4.
- Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344(10):732–8.
- Kamisawa T, Takuma K, Tabata T, Inaba Y, Egawa N, Tsuruta K, et al. Serum IgG4-negative autoimmune pancreatitis. J Gastroenterol. 2011;46(1):109–16.
- Yamaguchi Y, Kanetsuna Y, Honda K, Yamanaka N, Kawano M, Nagata M;Japanese study group on IgG4-related nephropathy. Characteristic tubulointerstitial nephritis in IgG4-related disease. Hum Pathol. 2012;43(4):536–49.
- Cornell LD. IgG4-related kidney disease. Curr Opin Nephrol Hypertens. 2012;21(3):279–88.
- Yoshita K, Kawano M, Mizushima I, Hara S, Ito Y, Imai N, et al. Light-microscopic characteristics of IgG4-related tubulointerstitial nephritis: distinction from non-IgG4-related tubulointerstitial nephritis. Nephrol Dial Transplant. 2012;27(7):2755–61.
- Kawano M, Mizushima I, Yamaguchi Y, Imai N, Nakashima H, Nishi S, et al. Immunohistochemical characteristics of IgG4-related tubulointerstitial nephritis: detailed analysis of 20 Japanese cases. Int J Rheumatol. 2012;2012:609795.
- Kawano M, Saeki T, Nakashima H, Nishi S, Yamaguchi Y, Hisano S, et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011:15(5):615–26.
- Raissian Y, Nasr SH, Larsen CP, Colvin RB, Smyrk TC, Takahashi N, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol. 2011;22(7):1343–52.
- Sato Y, Notohara K, Kojima M, Takata K, Masaki Y, Yoshino T. IgG4-related diseases: historical overview and pathology of hematological disorders. Pathol Int. 2010;60(4):247–58.
- Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012:22(1):21–30.
- Mizushima I, Yamada K, Fujii H, Inoue D, Umehara H, Yamagishi M, et al. Clinical and histological changes associated with corticosteroid therapy in IgG4-related tubulointerstitial nephritis. Mod Rheumatol. 2012;22(6):859–70.
- Chari ST, Takahashi N, Levy MJ, Smyrk TC, Clain JE, Pearson RK, et al. A diagnostic strategy to distinguish autoimmune pancreatitis from pancreatic cancer. Clin Gastroenterol Hepatol. 2009;7(10):1097–103.
- Matsubayashi H, Sawai H, Kimura H, Yamaguchi Y, Tanaka M, Kakushima N, et al. Characteristics of autoimmune pancreatitis based on serum IgG4 level. Dig Liver Dis. 2011;43(9):731–5.
- Zen Y, Nakanuma Y. IgG4-related disease: a cross-sectional study of 114 cases. Am J Surg Pathol. 2010;34(12):1812–9.
- Masaki Y, Kurose N, Umehara H. IgG4-related disease: a novel lymphoproliferative disorder discovered and established in Japan in the 21st century. J Clin Exp Hematopathol. 2011;51(1):13–20.
- Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25(9):1181–92.
- Makiishi T, Shirase T, Hieda N, Maeda S. Immunoglobulin G4-related disease with scant tissue IgG4. BMJ Case Rep. 2013 Jun 6;2013. doi:pii: bcr2013009800. 10.1136/bcr-2013-009800.
- Zen Y, Fujii T, Harada K, Kawano M, Yamada K, Takahira M, Nakanuma Y. Th2 and regulatory immune reactions are increased in immunoglobulin G4-related sclerosing pancreatitis and cholangitis. Hepatology. 2007:45(6):1538–46.
- Nakashima H, Miyake K, Moriyama M, Tanaka A, Watanabe M, Abe Y, et al. An amplification of IL-10 and TGF-beta in patients with IgG4-related tubulointerstitial nephritis. Clin Nephrol. 2010;73(5):385–91.
- Tanaka A, Moriyama M, Nakashima H, Miyake K, Hayashida JN, Maehara T, et al. Th2 and regulatory immune reactions contribute to IgG4 production and the initiation of Mikulicz disease. Arthritis Rheum. 2012;64(1):254–63.