
Abstract
Objective. To assess the long-term safety, immunogenicity, and efficacy of subcutaneous (SC) abatacept in combination with methotrexate (MTX) in Japanese patients with rheumatoid arthritis who were MTX inadequate responders, in a long-term extension (LTE) to a double-dummy, double-blind study (NCT01001832).
Methods. Patients, who had previously received SC or intravenous (IV) abatacept with MTX (6–8 mg/week) for 24 weeks, received SC abatacept (125 mg/week) with MTX for an additional 52 weeks. Safety, immunogenicity, and efficacy were assessed.
Results. The LTE included 112 patients. SC abatacept was generally well tolerated in the LTE, with no new safety signals. American College of Rheumatology 20, 50, and 70 response rates, disease activity score 28 (C-reactive protein) remission rates (< 2.6), and Health Assessment Questionnaire-Disability Index response rates (≥ 0.3 improvement from baseline) achieved at the end of the double-blind period were maintained over the LTE and were comparable in patients who received SC or IV abatacept in the double-blind period. Seropositivity for immunogenicity occurred in 4 (3.6%) patients. Self-injection of SC abatacept was well controlled and not associated with additional safety events.
Conclusions. SC abatacept had acceptable safety and was well tolerated and effective over the LTE (76 weeks in total), with low rates of immunogenicity in Japanese patients.
Introduction
Abatacept is a fully human, soluble fusion protein that selectively modulates the CD80/CD86:CD28 co-stimulatory signal required for full T-cell activation [Citation1]. Activated T-cells are implicated in the pathogenesis of rheumatoid arthritis (RA) [Citation2]. By competing with CD28 for CD80/CD86 binding, abatacept modulates serum levels of inflammatory cytokines and autoantibodies [Citation3].
Abatacept is available in intravenous (IV) and subcutaneous (SC) formulations. The efficacy and safety of SC abatacept in the treatment of RA, and its non-inferiority to IV abatacept, have been reported [Citation4–8]. Both IV and SC abatacept are approved in the USA, Europe, and Japan for the treatment of RA. In Japan, IV abatacept was approved for the treatment of RA in 2010 and SC abatacept was approved for the treatment of RA in 2013. The long-term safety profile of IV abatacept in Japanese patients with RA has been described previously [Citation9–12], but there is a lack of long-term safety and efficacy data for SC abatacept in this population.
The Abatacept Comparison of subQ versus intravenoUs in Inadequate Responders to mEthotrexate (ACQUIRE) study directly compared the efficacy and safety of IV and SC abatacept, with background methotrexate (MTX) [Citation4]. SC abatacept had comparable efficacy and safety to IV abatacept in patients with an inadequate response to MTX, as well as low immunogenicity, low rates of injection-site reactions, and a high 6-month retention rate. ACQUIRE did not include Japanese patients, and so a double-dummy, double-blind, Phase II/III bridging study with Japanese patients was conducted [Citation13]. Here we report the findings from a 1-year (52 weeks), open-label, long-term extension (LTE) of the Japanese bridging study with the aim of assessing the long-term safety, immunogenicity, and efficacy of SC abatacept plus MTX after 52 additional weeks of treatment.
Methods
Patient population
All patients who completed the 6-month, double-dummy, double-blind, Phase II/III bridging study were eligible to enter the open-label LTE period. The patient population comprised Japanese adults (age ≥ 20 years) with a diagnosis of active RA who had an inadequate response to MTX. Inclusion and exclusion criteria for the short-term bridging study were reported previously [Citation13]. All patients provided signed, written informed consent.
Study design
This study comprised a 6-month (24 weeks), multicenter, double-dummy, double-blind period followed by a 1-year (52 weeks) open-label LTE period, with an additional 6 months of follow-up (NCT01001832). The double-blind period was conducted across 34 sites in Japan between December 2009 and February 2011. During the double-blind period, patients were randomized (1:1) to SC abatacept (125 mg/week, IV loading of ∼10 mg/kg on Day 1) or IV abatacept (∼10 mg/kg, every 4 weeks), both with MTX (6–8 mg/week). In the LTE period, patients received SC abatacept (125 mg/week) with MTX for 52 weeks (Days 169–533); biweekly administration of SC abatacept was permitted only in patients with low body weight (≤ 50 kg). The MTX dose could be altered according to investigator discretion. Dose changes owing to adverse events (AEs) were prohibited, and non-biologic disease-modifying antirheumatic drugs were permitted. During the LTE period, SC abatacept was administered by self-injection (or by a caregiver), except where the investigator judged that injection at the study center was appropriate. Institutional Review Board/Independent Ethics Committee approval was received for the protocol and patient consent form, and the study was conducted in accordance with the Declaration of Helsinki [Citation14] and the International Conference on Harmonization Guideline for Good Clinical Practice [Citation15].
Assessments
Safety, immunogenicity, and efficacy were assessed in the intent-to-treat population during the LTE period. Safety assessments used data up to 8 weeks (56 days) post-treatment and included AE monitoring, physical examination, chest radiograph, electrocardiogram, physical measurements, breast cancer screening, vital signs, tuberculosis screening, and laboratory assessments. Blood samples were collected at Weeks 0, 12, 24, 36, 48, and 52 of the LTE period. Trough level serum abatacept concentration (Cmin), C-reactive protein (CRP), and immunogenicity were assessed at Weeks 12, 24, 36, 48, and 52 of the LTE period; and rheumatoid factor (RF) was assessed at Week 52. In patients who discontinued, immunogenicity sampling was performed at 7, 28, 84, and 168 days after the last dose of SC abatacept. Anti-abatacept immunogenicity testing was performed using a sensitive, validated electrochemiluminescence immunoassay method (Meso-Scale Discovery, Rockville, Maryland, USA). The electrochemiluminescence assay differentiated between two antibody specificities: immunoglobulin (Ig)G and/or junction region, and cytotoxic T-lymphocyte antigen-4 (CTLA-4) and possibly Ig. Neutralizing antibodies were assessed as described previously [Citation13]. Efficacy assessments included American College of Rheumatology (ACR) 20, 50, and 70 response rates over 533 days (the first administration of study drug in the double-blind period was Day 1); change in Disease Activity Score (DAS)28 (CRP) from baseline and the proportion of patients who achieved low disease activity (DAS28 < 3.2) and remission (DAS28 < 2.6) at Days 169 and 533; and change in Health Assessment Questionnaire-Disability Index (HAQ-DI) from baseline at Days 169 and 533 and the proportion of patients who achieved HAQ-DI response (reduction in HAQ-DI ≥ 0.3 units from baseline) over 533 days.
Sample size
Sample size calculations were based on Japanese guidelines for the assessment of RA drugs in clinical studies [Citation16] and were reported previously [Citation13].
Statistical analysis
No formal statistical tests were performed. Safety analyses included all treated patients in the LTE period, grouped according to the abatacept formulation received in the double-blind period. The evaluation of drug safety was based on AEs, vital signs, and laboratory abnormalities during the LTE period. Pharmacokinetic analyses included summarizing Cmin at Weeks 12, 24, 36, 48, and 52 using geometric means and coefficients of variation. RF and CRP were summarized by treatment received in the double-blind period. Immunogenicity was assessed by testing serum samples for the development of antibodies against abatacept. The incidence of positive response was summarized by treatment received in the double-blind period. Efficacy analyses included all patients who started the LTE period and received at least one dose of SC abatacept during the LTE period. ACR 20, 50, and 70 response rates; DAS28 (CRP) remission rates; and HAQ-DI response rates with exact 95% confidence intervals (CIs) were summarized at each time point by treatment received in the double-blind period. Missing values were not imputed.
Results
Patients
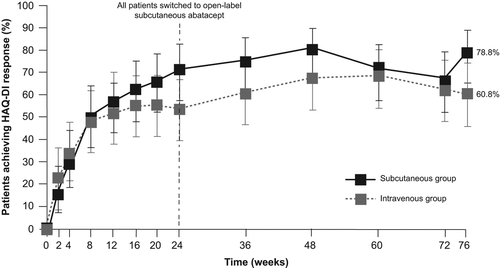
In the 6-month, double-blind period, 118 patients were randomized and treated with abatacept plus MTX. A total of 112 patients entered the LTE (during which all patients received 125 mg/week SC abatacept plus MTX): 56 who previously received SC abatacept (SC group) and 56 who previously received IV abatacept (IV group). The LTE period was completed by 52 (92.9%) patients in the SC group and 51 (91.1%) patients in the IV group ().
Figure 1. Patient flow. DB double-blind, IV intravenous, LTE long-term extension, SC subcutaneous.
In the LTE period, the mean (standard deviation [SD]) duration of exposure to abatacept was 13.5 (0.9) months in the SC group and 13.3 (1.8) months in the IV group (median [range]: 13.8 [Citation8–14] and 13.8 [Citation4–14] months, respectively). With regard to adherence, 41 (73.2%) patients in the SC group and 47 (83.9%) patients in the IV group received all the planned doses of SC abatacept; 7 (12.5%) and 3 (5.4%) patients missed one dose, 7 (12.5%) and 3 (5.4%) patients missed two doses, and 1 (1.8%) and 3 (5.4%) patients missed three or more doses, respectively. SC abatacept was administered by self-injection at least once in the LTE period by 105 (93.8%) patients. Baseline characteristics of patients were comparable with those for the 6-month, double-blind period [Citation13]. Characteristics were generally balanced in the SC and IV groups (); however, the SC group contained fewer women than the IV group (37 [66.1] vs. 46 [82.1%]), and patients in the SC group had longer mean (SD) disease duration (7.4 [8.8] vs. 5.3 [7.3] years) and lower mean (SD) CRP levels (1.90 [1.63] vs. 2.93 [2.79] mg/dL) than patients in the IV group. The mean (SD) weekly total MTX dose (expressed as mg/week) in the SC and IV groups, respectively, was 7.3 (1.0, n = 56) and 7.3 (0.9, n = 56) at baseline, 7.0 (1.7, n = 56) and 7.2 (1.2, n = 56) in the week including Day 169, and 7.7 (2.5, n = 52) and 7.4 (2.1, n = 51) in the week including Day 533.
Table 1. Patient and clinical disease characteristics at baseline.a
Safety
SC abatacept was generally well tolerated during the LTE period and the safety profile was consistent with that in the double-blind period (). During the LTE period, the long-term safety remained comparable between patients who had received SC and IV abatacept in the double-blind period. Serious adverse events (SAEs) were reported in 10 patients (); treatment-related SAEs were reported in 7 patients: dacryocystitis, bacterial pneumonia, interstitial lung disease, breast cancer, lower abdominal pain and pyrexia, colonic polyp and colon cancer, and dermal cyst. Three patients discontinued SC abatacept owing to AEs (includes discontinuations owing to SAEs): colon cancer (SAE), acquired dacryoadenitis and eyelid ptosis, and interstitial lung disease.
Table 2. Safety profile of abatacept in the double-blind period and in the open-label LTE period [Citation13].
Infections and infestations were reported in 58 (51.8%) patients; nasopharyngitis was the most frequently reported infection (28.6%). There were two serious infections: dacryocystitis and bacterial pneumonia. No opportunistic infections or autoimmune disorders were reported. Malignant neoplasms were reported in 2 patients during the LTE: colon cancer and breast cancer. Prespecified local injection-site reactions occurred in 2 patients; both cases were of mild severity and did not lead to discontinuation. Prespecified systemic injection reactions occurred in 5 patients; all cases were of mild or moderate severity and did not lead to discontinuation. There were no abnormalities in laboratory test values and vital signs. One patient died from B-cell lymphoma considered by the investigator to be related to the study drug. B-cell lymphoma occurred on Day 568, after the patient received his/her last dose of abatacept, and the patient died on Day 595. Overall, the long-term safety was similar in patients who received SC and IV abatacept in the double-blind period.
Immunogenicity
In the LTE period, seropositivity for anti-abatacept antibodies was detected in 2 (3.6%) patients from the SC group and in 2 (3.6%) patients from the IV group. Three patients demonstrated reactivity specific to the Ig and/or junction region, and 1 patient demonstrated reactivity specific to the CTLA-4 and possibly the Ig-specific region. Seropositivity for anti-abatacept antibodies did not appear to affect the efficacy or safety of abatacept. Rates of post-treatment immunogenicity were consistent with previous observation after a prolonged period of drug withdrawal [Citation17].
Following the LTE, there was a 6-month follow-up period during which patients continued to be monitored for immunogenicity. In the follow-up period, seropositivity for anti-abatacept antibodies was detected in 9 (20.0%) patients in the SC group (7 were newly detected in the follow-up period, and 2 were initially detected in the LTE period and continuously in the follow-up period), and in 4 (10.3%) patients in the IV group. Among the 13 patients who tested positive for anti-abatacept antibodies, 7 patients from the SC group and 3 patients from the IV group underwent a neutralizing antibody assay. Neutralizing antibodies were found in 1 patient in the IV group with reactivity specific to the CTLA-4 and possibly the Ig-specific region. The development of anti-abatacept antibodies was not associated with autoimmune disease or hypersensitivity, and the efficacy and pharmacokinetic profile of SC abatacept was unchanged in patients who were seropositive, relative to those who were seronegative.
Pharmacokinetics
During the LTE, the abatacept geometric mean Cmin was observed without stratification by the IV and SC groups, and remained consistent from Day 253 to Day 533, ranging from 33.34 to 39.09 μg/mL following SC administration. In patients who received weekly abatacept, the geometric mean Cmin was 36.33 μg/mL in patients with a body weight of < 60 kg (n = 58) compared with 27.52 μg/mL in patients with a body weight of ≥ 60 kg (n = 26). Abatacept Cmin decreased following a positive immunogenic response to abatacept in 1 patient from the IV group.
Efficacy
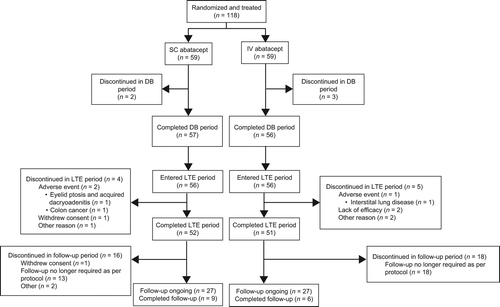
ACR 20, 50, and 70, DAS28 (CRP), and HAQ-DI response rates observed at the end of the double-blind period (Day 169) were at least maintained during the LTE, and showed a trend toward continued increases up to Day 533 in both the SC and the IV groups (). At Day 533 (Week 76), ACR 20, 50, and 70 response rates were 94.2, 78.8, and 57.7% in the SC group, and 96.1, 90.2, and 60.8% in the IV group, respectively ().
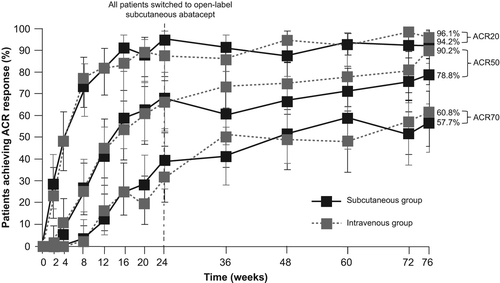
Figure 3. DAS28 (CRP) remission (< 2.6) rates at Weeks 24 and 76 (Weeks 24–76)a. Error bars represent 95% CIs. aAs-observed analysis.
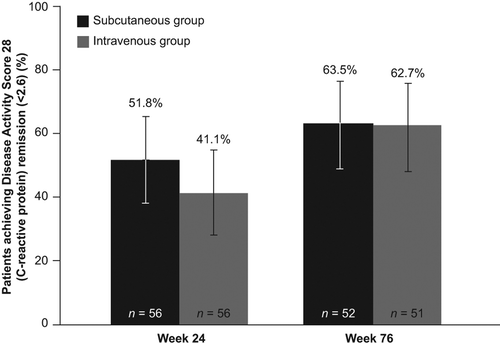
Figure 4. HAQ-DI response rates during the double-blind period (Weeks 0–24) and the open-label LTE period (Weeks 24–76)a. Error bars represent 95% CIs. aAs-observed analysis.
DAS28 (CRP) < 2.6 rates at Day 533 (Week 76) were 63.5% and 62.7% in the SC and IV groups, respectively (). The mean (95% CI) changes from baseline in DAS28 (CRP) at Day 169 (Week 24) and Day 533 (Week 76) were − 2.91 (− 3.17, − 2.65) and − 3.27 (− 3.58, − 2.97) in the SC group, and − 2.89 (− 3.23, − 2.54) and − 3.49 (− 3.82, − 3.17) in the IV group, respectively. The percentages (95% CI) of patients with low disease activity at Day 169 (Week 24) and Day 533 (Week 76) were 69.6% (55.9, 81.2) and 86.5% (74.2, 94.4) in the SC group, and 67.9% (54.0, 79.7) and 82.4% (69.1, 91.6) in the IV group, respectively.
At Day 533 (Week 76), HAQ-DI response rates were 78.8% in the SC group and 60.8% in the IV group (). Mean (95% CI) changes in HAQ-DI score from baseline at Day 169 (Week 24) and Day 533 (Week 76) were − 0.60 (− 0.74, − 0.47) and − 0.71 (− 0.87, − 0.55) in the SC group, and − 0.63 (− 0.78, − 0.49) and − 0.71 (− 0.89, − 0.52) in the IV group, respectively.
At baseline, 48 subjects in the SC group and 47 subjects in the IV group tested positive for RF (defined as RF > 20 U/mL). Of these patients, conversion to seronegativity for RF was confirmed on Day 533 (Week 76) in 7 (14.6%) patients in the SC group and 4 (8.5%) patients in the IV group. Of the 7 patients in the SC group and 8 patients in the IV group who were RF negative at baseline, 1 subject in the IV group converted to seropositivity for RF on Day 533 (Week 76).
Results in patients with low body weight (≤ 50 kg)
A total of 41 patients in the LTE period had a body weight ≤ 50 kg at baseline, and 2 patients in this subgroup discontinued from the LTE period (patient relocation and AE). During the 1-year LTE period, 23/41 (56.1%) patients with body weight ≤ 50 kg switched from weekly dosing of 125-mg abatacept to biweekly dosing; 15 patients were from the IV group and 8 were from the SC group. Of the 23 patients who switched to biweekly dosing of abatacept, 3 patients had a clinically meaningful increase in DAS28 (CRP) of ≥ 0.6 but did not switch back to weekly dosing, and 7 patients switched back to weekly dosing based on the investigator's judgment. The efficacy of abatacept (measured by DAS28 [CRP]) was maintained during the LTE period in the 18 patients with body weight ≤ 50 kg who did not switch to biweekly dosing. Temporary clinically meaningful increases in DAS28 (CRP) of ≥ 0.6 occurred in 3/18 patients with body weight ≤ 50 kg who did not switch to biweekly dosing; these increases in DAS28 (CRP) were normalized within 2 visits.
Discussion
The present study assessed the long-term safety and efficacy of weekly SC abatacept plus MTX in Japanese patients who were MTX inadequate responders in an LTE of the 24-week, double-blind ACQUIRE bridging study. Treatment with SC abatacept over 1 year in the LTE was associated with high patient retention; the majority of patients (92.9% in the SC abatacept group and 91.1% in the IV abatacept group) completed the LTE and there were only three discontinuations, all due to AEs. This high patient retention reduces selection bias due to patient attrition. Overall, the efficacy and safety profile of SC abatacept in Japanese patients was consistent with that observed in the global population [Citation4–7,Citation8].
No new abatacept safety signals were identified, and the safety profile of SC abatacept in the LTE was consistent with findings in the double-blind period [Citation13], as well as with post-marketing surveillance data in Japanese patients [Citation9]. During the LTE, rates of discontinuation owing to AEs and SAEs were low (0.9 and 2.7%, respectively). SC administration of biologics can be associated with injection-site reactions and injection-site pain [Citation18,Citation19]; however, there was a low incidence of local injection-site reactions in this study (1.8%). Long-term exposure to abatacept did not appear to result in an increased incidence of safety events, a finding that was consistent with safety results from an integrated analysis of five clinical trials involving SC abatacept, with patient exposure of up to 4.5 years [Citation20,Citation21].
Biologic therapies have the potential to elicit an immune response, and the development of immunogenicity could reduce clinical response to abatacept in patients with RA [Citation22]. In agreement with previous findings, a low number of patients had an immune response to abatacept (3.6% of patients, in both the SC and IV abatacept groups) [Citation6].
Abatacept serum trough concentrations were maintained through continuous long-term therapy with weekly SC dosing of abatacept 125 mg, but were found to be higher in patients with body weight < 60 kg versus ≥ 60 kg. Peak serum abatacept concentrations (Cmax) in the IV and SC groups (n = 29 and n = 30, respectively) during the 24-week, double-blind period were 277.4 and 42.6 μg/mL, respectively [Citation13]. The Cmax of SC abatacept (125 mg/week) in the LTE was not obtained in this study, as blood samples were collected just prior to SC abatacept administration and not at the necessary time points required to calculate the Cmax.
ACR 20, 50, and 70 response rates; DAS28 (CRP) remission rates; and HAQ-DI response rates were similar in the SC and IV groups (as shown by overlapping 95% CIs) and were maintained from the end of the double-blind period to the end of the LTE period. SC abatacept was highly effective in achieving both clinical and functional improvements, even with a lower mean dose of concomitant MTX compared with international clinical trials (this may be due to the lower body weight of patients in this study) [Citation4,Citation6,Citation7].
SC abatacept was injected by an investigator at the site in the double-blind period, and was self-injected by a patient or caregiver in the LTE period. After the introduction of self-injection to the study, the efficacy achieved in the double-blind period was maintained during the LTE period, and there were no new safety signals related to self-injection. These findings indicate that the self-administration of SC abatacept was a viable alternative to IV administration in Japanese patients with RA and inadequate response to MTX. Further studies are required to better understand the long-term efficacy, safety, and adherence to abatacept in a real-world setting in Japan.
A limited number of patients with body weight ≤ 50 kg switched from weekly to biweekly dosing in the LTE period (23/41 patients), and 10 patients who switched to biweekly dosing showed a clinically meaningful increase in DAS28 (CRP). Limited conclusions can be drawn from the data set; however, there is a suggestion that biweekly dosing of 125-mg SC abatacept may be inadequate to maintain efficacy in Japanese patients with RA and a body weight of ≤ 50 kg.
The safety and efficacy of SC abatacept beyond the duration of the LTE period (76 weeks of abatacept in total) is unknown in Japanese patients with RA. For chronic diseases such as RA, longer term safety data are important. Radiographic progression was not evaluated in this study.
In conclusion, the safety profile of SC abatacept during the LTE period was similar to that reported during the double-blind period. There were no new safety signals. The rate of immunogenicity was low in the LTE period. Immunogenicity did not appear to affect the safety, pharmacokinetics, or efficacy of abatacept. Rates of immunogenicity for SC abatacept in the LTE period (on-treatment [1 year] and post-treatment [6 months]) were similar to those historically found with SC abatacept. Improvements in RA signs and symptoms and physical function that were achieved in 6 months with IV or SC abatacept were maintained with SC abatacept through an additional 1-year period. The consistent long-term safety and maintained clinical efficacy support the use of SC abatacept as a treatment option for Japanese patients with RA who are MTX inadequate responders. The self-injection of SC abatacept was accepted and well controlled in Japanese patients with RA and was not associated with additional safety events.
Acknowledgments
The authors would like to thank the investigators and medical personnel involved in this study at the following sites in Japan: Hokkaido University Hospital, Hokkaido; Sapporo City General Hospital, Hokkaido; Taga General Hospital, Ibaraki; National Hospital Organization Utsunomiya National Hospital, Tochigi; Jichi Medical University Hospital, Tochigi; Gunma University Hospital, Gunma; Saitama Medical University Hospital, Saitama; Saitama Medical Center, Saitama; Keio University Hospital, Tokyo; Tokyo Medical & Dental University Hospital Faculty of Medicine, Tokyo; Yokohama Rosai Hospital, Kanagawa; National Hospital Organization Sagamihara National Hospital, Kanagawa; Nagano Red Cross Hospital, Nagano; Seirei Hamamatsu General Hospital, Shizuoka; Kanzaki Municipal General Hospital, Hyogo; Matsubara Mayflower Hospital, Hyogo; Higashi-Hiroshima Memorial Hospital, Hiroshima; National Hospital Organization Kyushu Medical Center, Fukuoka; University of Occupational and Environmental Health Hospital, Fukuoka; National Hospital Organization Hokkaido Medical Center, Hokkaido; Inoue Hospital, Gunma; Kitasato Institute Medical Center Hospital, Saitama; Japanese Red Cross Narita Hospital, Chiba; Tokyo Metropolitan Police Hospital, Tokyo; Yokohama Minami Kyousai Hospital, Kanagawa; Osaka Rehabilitation Hospital, Osaka; Medical Corporation Matsubara Clinic, Hyogo; Kurashiki Kosai Hospital, Okayama; St. Mary's Hospital, Fukuoka; Hokkaido Medical Center for Rheumatic Diseases, Hokkaido; Shizuoka Kosei General Hospital, Shizuoka; Izumihara Rheumatoid Arthritis and Internal Medicine Clinic, Kagoshima; and Hirose Clinic, Saitama.
Professional medical writing and editorial assistance was provided by Stephen Moore PhD at Caudex Medical and was funded by Bristol-Myers K.K.
All authors participated in the development of this manuscript, provided approval of the final version of the manuscript submitted for publication, and were either study investigators (K. Amano, T. Matsubara, H. Inoue, M. Iwahashi, T. Kanamono, T. Nakano, S. Uchimura, T. Izumihara, and T. Takeuchi) or were involved in the study design/conception and data interpretation (A. Yamazaki and C.S. Karyekar) of study NCT01001832.
Conflicts of interest
This study was sponsored by Bristol-Myers Squibb.
K. Amano has received lecture fees from AbbVie GK., Astellas Pharma Inc., Bristol-Myers K.K., Chugai Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Eisai Co., Ltd., and Pfizer Japan Inc., and has received research grants from Astellas Pharma Inc., Bristol-Myers K.K., Chugai Pharmaceutical Co., Ltd., and Mitsubishi Tanabe Pharma Corporation.
T. Matsubara has received lecture fees from Pfizer Japan Inc., Janssen Pharmaceutical Co., Ltd., and Astellas Pharma Inc.; and research grants from Quintiles Transnational Japan K.K., Janssen Pharmaceutical Co., Ltd., Takeda Chemical Industries, Ltd., Daiichi Sankyo Co., Ltd., Astellas Pharma Inc., Eli Lilly Japan K.K., MSD Co., Ltd., Nippon Kayaku Co., Ltd., Parexel International Corp., Pfizer Japan Inc., and Bristol-Myers Squibb.
M. Iwahashi has received lecture fees from Bristol-Myers K.K., Chugai Pharmaceutical Co., Ltd., Pfizer Japan Inc., Mitsubishi Tanabe Pharma Co., Janssen Pharmaceutical K.K., Abbott Japan Co., Ltd., Astellas Pharma Inc., Santen Pharmaceutical Co., Ltd., and Asahi Kasei Pharma Co.
T. Nakano has received lecture fees from Eisai Co., Ltd., Pfizer Japan Inc., Abbott Japan Co., Ltd., Daiichi Sankyo Company, Ltd., Astellas Pharma Inc., Ono Pharmaceutical Co., Ltd., UCB Japan Co. Ltd., Mitsubishi Tanabe Pharma Co., Asahi Kasei Pharma Co., and Chugai Pharmaceutical Co., Ltd.
A. Yamazaki is a paid employee of Bristol-Myers K.K.
C.S. Karyekar is a paid employee of Bristol-Myers Squibb and owns Bristol-Myers Squibb stock.
T. Takeuchi has received grants from Astellas Pharma Inc., Bristol-Myers K.K., Chugai Pharmaceutical Co., Ltd., Daiichi Sankyo Co., Ltd., Eisai Co., Ltd., Mitsubishi Tanabe Pharma Co., Pfizer Japan Inc., Santen Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co. Ltd., Teijin Pharma Ltd., AbbVie GK., Asahi Kasei Pharma Co., Taisho Toyama Pharmaceutical Co., Ltd., and SymBio Pharmaceuticals Ltd.; lecture fees from AbbVie GK., Ltd., Bristol-Myers K.K., Chugai Pharmaceutical Co., Ltd., Eisai Co., Ltd., Janssen Pharmaceutical K.K., Mitsubishi Tanabe Pharma Co., Pfizer Japan Inc., Takeda Pharmaceutical Co. Ltd., Astellas Pharma Inc., and Daiichi Sankyo Co., Ltd.; and consulting fees from AstraZeneca K.K., Eli Lilly Japan K.K., Novartis Pharma K.K., Mitsubishi Tanabe Pharma Co., Asahi Kasei Medical K.K., AbbVie GK, Daiichi Sankyo Co., Ltd., and Bristol-Myers K.K.
T. Tanaka, H. Inoue, T. Kanamono, S. Uchimura, and T. Izumihara have nothing to disclose.
References
- Moreland LW, Alten R, Van den Bosch F, Appelboom T, Leon M, Emery P, et al. Costimulatory blockade in patients with rheumatoid arthritis: a pilot, dose-finding, double-blind, placebo-controlled clinical trial evaluating CTLA-4Ig and LEA29Y eighty-five days after the first infusion. Arthritis Rheum. 2002;46(6):1470–9.
- Cope AP, Schulze-Koops H, Aringer M. The central role of T cells in rheumatoid arthritis. Clin Exp Rheumatol. 2007;25(5 Suppl 46): S4–11.
- Cutolo M, Nadler S. Advances in CTLA-4-Ig-mediated modulation of inflammatory cell and immune response activation in rheumatoid arthritis. Autoimmun Rev. 2013;12(7):758–67.
- Genovese MC, Covarrubias A, Leon G, Mysler E, Keiserman M, Valente R, et al. Subcutaneous abatacept versus intravenous abatacept: a phase IIIb noninferiority study in patients with an inadequate response to methotrexate. Arthritis Rheum. 2011;63(10):2854–64.
- Keystone EC, Kremer JM, Russell A, Box J, Abud-Mendoza C, Elizondo MG, et al. Abatacept in subjects who switch from intravenous to subcutaneous therapy: results from the phase IIIb ATTUNE study. Ann Rheum Dis. 2012;71(6):857–61.
- Nash P, Nayiager S, Genovese MC, Kivitz AJ, Oelke K, Ludivico C, et al. Immunogenicity, safety, and efficacy of abatacept administered subcutaneously with or without background methotrexate in patients with rheumatoid arthritis: results from a phase III, international, multicenter, parallel-arm, open-label study. Arthritis Care Res (Hoboken). 2013;65(5):718–28.
- Weinblatt ME, Schiff M, Valente R, van der Heijde D, Citera G, Zhao C, Maldonado M, Fleischmann R. Head-to-head comparison of subcutaneous abatacept versus adalimumab for rheumatoid arthritis: Findings of a phase IIIb, multinational, prospective, randomized study. Arthritis Rheum. 2013;65(1):28–38.
- Schiff M, Weinblatt ME, Valente R, van der HD, Citera G, Elegbe A, Maldonado M, Fleischmann R. Head-to-head comparison of subcutaneous abatacept versus adalimumab for rheumatoid arthritis: two-year efficacy and safety findings from AMPLE trial. Ann Rheum Dis. 2013;73:86–94.
- Koike T, Harigai M, Ishiguro N, Inokuma S, Ryu J, Takei S, et al. Safety and effectiveness of abatacept in 3985 Japanese patients with rheumatoid arthritis; Japan all-cases post-marketing surveillance. Arthritis Rheum. 2013;65(Suppl 10):1415.
- Matsubara T, Inoue H, Iwahashi M, Yamazaki A, Takeuchi T, Japan Abatacept Study Group. A multi-center, double-dummy, double-blind study of subcutaneous (SC) abatacept (ABA) compared with intravenous (IV) ABA in Japanese rheumatoid arthritis patients with inadequate response to methotrexate [abstract]. Ann Rheum Dis. 2012;71(Supp l3):197.
- Matsubara T, Yamana S, Tohma S, Takeuchi T, Kondo H, Kohsaka H, et al. Tolerability and efficacy of abatacept in Japanese patients with rheumatoid arthritis: a phase I study. Mod Rheumatol. 2013; 23(4):634–45.
- Takeuchi T, Matsubara T, Nitobe T, Suematsu E, Ohta S, Honjo S, et al. Phase II dose-response study of abatacept in Japanese patients with active rheumatoid arthritis with an inadequate response to methotrexate. Mod Rheumatol. 2013;23(2):226–35.
- Iwahashi M, Inoue H, Matsubara T, Tanaka T, Amano K, Kanamono T, et al. Efficacy, safety, pharmacokinetics, and immunogenicity of abatacept administered subcutaneously or intravenously in Japanese patients with rheumatoid arthritis and inadequate response to methotrexate: a Phase II/III, randomized study. Mod Rheumatol. 2014;24:885–91.
- World Medical Association Declaration of Helsinki. Recommendations guiding physicians in biomedical research involving human subjects. JAMA. 1997;277:925–6.
- ICH Harmonised Tripartite Guideline. Guideline for Good Clinical Practice. J Postgrad Med. 2001;47:199–203.
- Evaluation and Licensing Division Pharmaceutical and Food Safety Bureau Ministry of Health Labour and Welfare. Guideline for Clinical Evaluation of Anti-rheumatic Drugs. Available from: http://www.pref.chiba.lg.jp/yakumu/iyakubugaihin/documents/rgg.pdf (accessed 27 November 2014).
- Kaine J, Gladstein G, Strusberg I, Robles M, Louw I, Gujrathi S, et al. Evaluation of abatacept administered subcutaneously in adults with active rheumatoid arthritis: impact of withdrawal and reintroduction on immunogenicity, efficacy and safety (phase IIIb ALLOW study). Ann Rheum Dis. 2012;71(1):38–44.
- Humira Summary of Product Characteristics. Available from: http://www.medicines.org.uk/EMC/medicine/21201/SPC/Humira+ Pre-filled+ Pen%2c+ Pre-filled+ Syringe+ and+ Vial/ (accessed 27 November 2014).
- Frenken LA, van Lier HJ, Gerlag PG, den HM, Koene RA. Assessment of pain after subcutaneous injection of erythropoietin in patients receiving haemodialysis. BMJ. 1991;303(6797):288.
- Alten R, Kaine J, Keystone EC, Nash P, Delaet I, Qi K, et al. Safety of subcutaneous abatacept in patients with rheumatoid arthritis (RA): Integrated analysis of five clinical trials up to 4.5 years. Ann Rheum Dis. 2011;70 (Suppl 3):617.
- Alten R, Kaine J, Keystone EC, Nash P, Delaet I, Qi K, Genovese M. Safety profile of subcutaneous abatacept focusing on clinically relevant events in patients with rheumatoid arthritis (RA) and up to 4.5 years of exposure. Arthritis Rheum. 2011;63 (Suppl 10):S150–1.
- Schellekens H. The immunogenicity of therapeutic proteins. Discov Med. 2010;9(49):560–4.