Abstract
A 3D-QSAR investigation of 95 diaminobenzophenone yeast farnesyltransferase (FT) inhibitors selected from the work of Schlitzer et al. showed that steric, electrostatic, and hydrophobic properties play key roles in the bioactivity of the series. A CoMFA/CoMSIA combined model using the steric and electrostatic fields of CoMFA together with the hydrophobic field of CoMSIA showed significant improvement in prediction compared with the CoMFA steric and electrostatic fields model. The similarity of the 3D-QSAR field maps for yeast FT inhibition activity (from this work) and for antimalarial activity data (from previous work) and the correlation between those activities are discussed.
Introduction
Human malaria, mainly caused by the apicomplexan parasite Plasmodium falciparum, is one of the most important infectious diseases in the world, leading to 300 million cases and 1.5–2.7 million deaths per yearCitation1,Citation2. Existing antimalarial chemotherapy agents include quinolines, antifolates, atovaquone/proguanil, artemisinins, and some more general antibioticsCitation3,Citation4. Just like for many other infectious diseases, most of the existing drugs are facing emerging resistance by P. falciparum, and some of the drugs such as artemisinins and atovaquone/proguanil are also very costly. Thus there is a great need for novel and inexpensive antimalarial compoundsCitation5–9.
One of the promising antimalarial targets is inhibition of protein farnesyltransferase (FT)Citation10,Citation11. The heterodimeric zinc-containing FT has been pegged as an anticancer target since it was discovered that it farnesylates Ras, which is overexpressed in certain kinds of malignant tumorsCitation12–15. More recently, however, evidence has shown that there must be another, yet to be discovered, mechanism of anticancer action since FT inhibitors also kill tumor cells which do not overexpress Ras, so currently the mechanism of action is unknownCitation12–15. Several FT inhibitors have proceeded into clinical trials for cancer chemotherapyCitation12,Citation13. Meanwhile, in the last few years, several groups have found that application of FT inhibitors to cells infected with P. falciparum led to a decrease in farnesylated proteins and to the associated lysis of the parasitesCitation16–18. The FT inhibitors were selectively toxic against the parasite as compared to against mammalian cells.
As part of our continuing effort to find novel lead compounds for antimalarial drugs, we selected for analysis 95 compounds based on a 2,5-diaminobenzophenone scaffold (), from the work of Schlitzer et al., who reported inhibition activity data against yeast FTCitation19–24. We performed a three-dimensional quantitative structure–activity relationship (QSAR) investigation, using the CoMFA (comparative molecular field analysis) and CoMSIA (comparative molecular similarity indices analysis) methods for the yeast FT activity data, in order to gain understanding of what factors govern the interaction between this series of compounds and FT. CoMFA and CoMSIA are commonly used 3D-QSAR methods. Based on the assumption that all the investigated compounds adopt the same binding mode with the enzyme, the CoMFA method calculates steric and electrostatic properties according to Lennard–Jones and Coulomb potentials in the space surrounding each of the properly aligned molecules in the data setCitation25. The CoMSIA approach calculates similarity indices, including steric occupancy, electrostatic field, local hydrophobicity, hydrogen-bond donor, and hydrogen-bond acceptor fields, under the same conditionsCitation26. After calculating these fields for every properly aligned structure, partial least squares (PLS) is used to correlate these properties to the bioactivity of the compounds. 3D-QSAR has previously been applied to other FT inhibitor classesCitation27–31 but not to the FT inhibition activities of the diaminobenzophenones we consider in this work. We previously preparedCitation32 3D-QSAR models for 2,5-diaminobenzophenones, but used in vitro antimalarial activity data (P. falciparum growth inhibition) rather than the yeast FT inhibition activity of this work.
Table 1. Structure and experimental yeast farnesyltransferase inhibitory activity of the data set (test set in bold).
Materials and methods
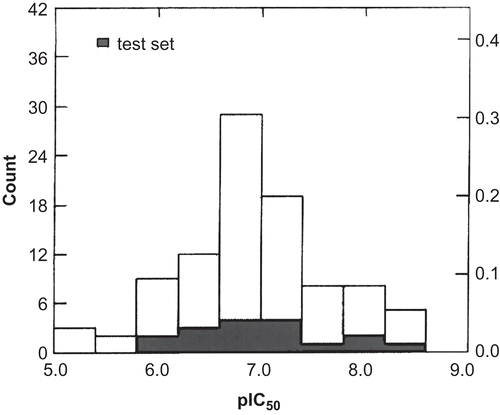
shows the structures of the 95 compounds used in this study and their observed anti-yeast farnesyltransferase activities (pIC50). The data set we used included 31 N-(4-tolylacetylamino-3-benzoylphenyl)-3-arylfurylacrylic acid amides, 22 N-(4-acylamino-3-benzoylphenyl)-3-[5-(4-nitrophenyl)-2-furyl]acrylic acid amides, 14 N-(4-acylamino-3-benzoylphenyl)-4-nitrocinnamic acid amides, four N-(4-aminoacylamino-benzoylphenyl)-3-[5-(4-nitrophenyl)-2-furyl] acrylic acid amides, eight 5-arylacryloylaminobenzophenones, and 16 2-(aminoacylamino)benzophenones. The compounds and their experimental activities (IC50) against yeast FT all originated from the same laboratoryCitation19–24. shows the distribution of pIC50 for the whole data set, ranging from 5.00 to 8.40 with a variance of 0.709.
Figure 1. Distribution of pIC50 for the data set.
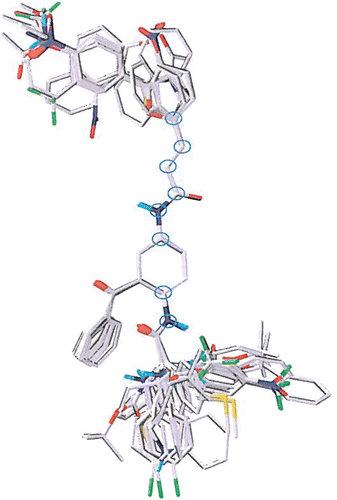
Molecular modeling, CoMFA and CoMSIA, were performed using Sybyl 7.2Citation33. The putative bioactive conformation was the minimum energy conformation of 43 (see reference 19). Each compound in the data set was minimized, adjusted manually to approximate the template conformation, and then minimized again with the MMFF94 force field to obtain the final geometry used for 3D-QSAR. Then the minimized conformations were aligned to the template using eight reference atoms in the backbone of the 2,5-diaminobenzophenone scaffold. shows the reference atoms and alignment. Initially the whole aligned data set was subjected to CoMFA and CoMSIA analysis, but compounds 4, 5, 18, 27, 35, 52, 78, 85, and 89 were identified as outliers by Grubbs’ test in both casesCitation34. After the nine outliers were omitted, the remaining compounds were divided into a 69 compound training set and 17 compound test set (bold in ), choosing a matching number of compounds from each activity range and from each compound class for each set (see ).
Figure 2. Structure alignment of the whole data set. The eight common backbone atoms used for alignment are circled.
For CoMFA and CoMSIA field calculations, a 3D cubic lattice with 2.0 å grid spacing was created to surround the aligned moleculesCitation25,Citation26. CoMFACitation25 fields were calculated using a sp3-carbon probe atom with a van der Waals radius of 1.52 å and a charge of +1.0 to generate steric (Lennard–Jones 6–12 potential) and electrostatic (Coulombic potential) fields with a distance-dependent dielectric at each lattice point and an energy cutoff of 30 kcal · mol−1. CoMSIA calculates similarity indices at the lattice pointsCitation26. Five physicochemical properties (steric, electrostatic, hydrophobic, and hydrogen-bond donor and acceptor) were evaluated using the similarity indices. Settings used included: probe atom radius = 1 å; charge, hydrophobicity, and hydrogen bond donating and accepting factors each set to 1; and attenuation factor α = 0.3Citation26.
CoMFA or CoMSIA descriptors were used as independent variables, and pIC50 values were the dependent variables in PLS analysis to derive QSAR models. The statistical quality of the models was evaluated using r2, q2, and r2pred, which are defined using sums of squares of the residuals between calculated and experimental activities (RSS) and of the differences between experimental activities and their mean (TSS) as 1 − (RSS/TSS). The sums are over training set compounds for r2 and q2 and over test set compounds for r2pred. The calculated activities are those from the QSAR model itself for r2 and r2pred, whereas for q2 they are instead those predicted by successively leaving out one training set compoundCitation32. The SAMPLS (sample-distance PLS) algorithm was used to determine the optimal number of components (N, given in ), based on how much q2 increased as N was increased. The optimal number of components was then used to derive the final QSAR model. The predictive power was tested by calculating r2pred for all models which had q2 > 0.3. The variance was calculated as RSS/n, for n compounds. For each model we again used Grubbs’ test to check for outliersCitation34.
Table 2. Statistical parameters of CoMFA (1–3), CoMSIA (4–8), and combined CoMFA/CoMSIA (9) models.
For each CoMFA or CoMSIA model, figures depicting coefficient contour maps for particular fields—in which the coefficients are scaled by the standard deviation in order to be on a percent scale—give insight into regions around the aligned ligands in which functional group modifications will lead to improved or worsened activity. In this work, such properties are depicted for regions having scaled coefficients either >80% (favored) or <20% (disfavored)Citation25.
Results
By thorough study we found that steric, electrostatic, and hydrophobic features are key factors that impact the bioactivity of this series of compounds, whereas the hydrogen bond donor and acceptor fields have no significant correlation with yeast FT inhibition activity. The same three fields were found to be most important in our previous study on the in vitro antimalarial activity of a set of 92 2,5-diaminobenzophenones, which included 60 of the same molecules as in this workCitation32.
summarizes the full investigation based on the training set of 69 compounds. For the CoMSIA models, statistical parameters showed significance for steric, electrostatic, and hydrophobic features, but hydrogen-bond donor or acceptor fields made insignificant contributions to the models and hence such features are predicted not to influence the activity of the compounds. For steric and electrostatic properties, the CoMFA fields were far more interpretive than the CoMSIA fields; this may be due to the high structural diversity of the data set. Hydrophobicity (model 6) was the most interpretive factor for this series of compounds (see reference 32). With q2 = 0.416 and r2pred = 0.370, model 6 had the strongest predictive power among the single field models (models 1, 2, 4, 5, and 6). Though the CoMFA electrostatic field (model 2) showed similar or even better q2 (0.414), r2 (0.711), and F (53.25) than model 6 (0.416, 0.672, and 44.36, respectively), its very low r2pred (0.172) showed that model 2 is not as reliable as models 3 (r2pred = 0.383), 6 (r2pred = 0.370), and 9 (r2pred = 0.495). In order to take full advantage of both CoMFA and CoMSIA fields, we made a combined CoMFA and CoMSIA model (model 9), using the steric and electrostatic field of CoMFA plus the hydrophobic field of CoMSIA. This model showed significant improvement in prediction compared with separate CoMFA or CoMSIA models (). This CoMFA plus CoMSIA model is the one we used for graphical analysis below. shows the residuals of the test set predictions by all models.
Table 3. Residualsa of test set predictions for 3D-QSAR models.
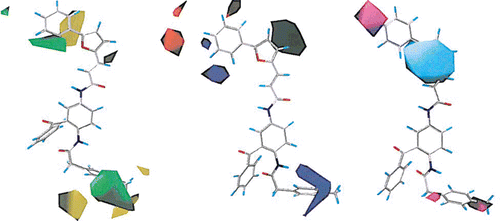
The contribution maps from the combined CoMFA and CoMSIA model (model 9) shown in illustrate how different fields affect the bioactivity of these compounds, with the template compound as reference. These contour maps are basically consistent with those depicted in our previous paperCitation32 on antimalarial activity of these compounds. For the steric field, the main difference between the contour maps was at the upper part around the furan ring; this is because some of the structures analyzed in this work contain totally different features compared to those studied in our previous antimalarial paperCitation32, especially in the cinnamic acid amide derivative cluster (compounds 74–88) and other structures without a furan ring in that position (compounds 89–95). The structural variants are shown clearly in . Because compounds 88 and 90–94 all have low activities (pIC50 = 5–6), the big and small blocks of yellow around the furan ring indicate that in that area, larger substituent groups (mainly larger rings) will result in activity loss. In the electrostatic contour maps, the differences are also caused by the same compounds 74–95. The activity trend for 88, 91, 94, and 95 indicates that besides the negative steric effect on the activity of these structures, electronegative atoms /groups on the first ring of the multi-ring R group (see ) will enhance the activity of these compounds. The hydrophobic contour map of this work compared to the previous workCitation32 shows a difference only at the lower left part of the picture: while for the previous antimalarial model there was a small hydrophobic block and a smaller hydrophilic block indicating the modification preferences of structures such as 34 and 58–65, no information of this kind was produced by this study of FT inhibition. This may be attributed to several structurally different compounds with available FT inhibition data but not antimalarial data, such as 66–73, distorting the field calculation in this region.
Figure 3. Contour (stdev*coeff) field maps for model 9, shown around the template compound. Left: Steric map: the green (yellow) region shows where steric bulk increases (decreases) activity. Center: Electrostatic map: the blue (red) region shows where electronegative (electropositive) groups increase the activity. Right: Hydrophobic map: the magenta (cyan) region shows where hydrophobic groups increase (decrease) the activity.
Discussion
The mechanism of antimalarial activity of a representative benzophenone was determined to be protein farnesyltransferase inhibition in P. falciparum in an experimental study by Wiesner et al.Citation18, which included a marked inhibition of farnesylation at 10 nM. Since all the benzophenones used in our 3D-QSAR analysis are structurally similar to the benzophenone from reference 18 and have reported FT inhibitory activity in yeast (pIC50 range of 5–8.4), it can be hypothesized that for all the antimalarial benzophenones, FT inhibition is their major mode of activity against P. falciparum. As one check of the validity of this hypothesis, attempts have been made to see if there is a correlation between antimalarial activity and yeast FT inhibition. In such work, yeast FT was used as a convenient surrogate for what would be more useful, Plasmodium falciparum FT (Pf FT).
In one such attempt by Fucik et al.Citation35, the antimalarial and yeast FT inhibition activities of benzophenones were analyzed and found not to be correlated. Fucik et al. did this analysis only for 11 of the 19 compounds in their paper, excluding eight cinnamic acid amide derivatives which exhibited poor solubility that would limit in vitro antimalarial activity. They suggested two possible reasons why the two activities were not correlated: first, the permeation requirements are different in the two assays (the antimalarial assay involves cultured whole parasite cells, whereas for FT binding the isolated enzyme is used); and second, the amino acid sequences are similar but not the same in the active sites of yeast and Pf FT.
Our analysis of the 11 compounds used to check the correlation by Fucik et al.Citation35 shows that indeed permeation may play a crucial role in the antimalarial assay, whereas it would not have an influence in the isolated enzyme assay. This may be the reason why a correlation between the two activities was not obtained by Fucik et al. Their most active antimalarial compound 4e, with a hydrophobic –CF3 substituent, has the highest Clog P (calculated log P) of 8.76, whereas the least active compound 4b, with a polar –OH substituent, has a significantly lower Clog P of 7.21. The more polar groups such as –OH and –NO2 (IC50: 3250 and 1400 nM, respectively) limit the permeation of these compounds into the parasites, whereas hydrophobic groups such as –CF3 and –Cl (IC50: 61 and 125 nM, respectively) facilitate the permeation. Hence, the poor correlation between the activities of these 11 compounds may be because of differential permeation in the two assays.
In another attempt to check whether antimalarial and FT inhibition activities are correlated, Wiesner et al.Citation36 studied eight other benzophenones, and found no correlation. The authors attributed this to FT species difference and limited membrane penetration. Our analysis of their data set shows that their compounds 1a, 1b, and 1f, with increasing hydrophobicity (Clog P = 6.80, 7.97, and 8.73, respectively), showed nearly identical antimalarial activities (IC50 = 5.7–5.9 μM). Interestingly, compound 1e, with a polar piperonyl replacement for the hydrophobic naphthyl ring found in compound 1b, also showed the same antimalarial activity (IC50 = 5.8 vs. 5.7 μM), but markedly different enzyme binding (IC50 = 115 vs. 6300 nM, respectively). This suggests that the membrane permeation of the compounds in that work is of limited concern with respect to their antimalarial activity, whereas the active site amino acid sequence difference between yeast and Pf FT is of major concern. This is especially likely because the compounds in reference 36 differ in substitution in the arylfuryl general core of benzophenones, which occupies the far aryl binding site in FTCitation23. Homology models of yeastCitation23 and P. falciparumCitation21 FT were built by the original authors who reported these benzophenones. Even though overall the active site amino acids are nearly identical, there are some subtle differences between the FTs of the two species that may influence the ligand binding. Notably, key amino acids found in the vicinity of the far aryl binding pocket in yeast FT (Ala47, Arg48, and Asp43) are different in P. falciparum FT (Thr47, Pro48, and Ile43), which can affect sterics and other specific interactions of the benzophenones with the active site.
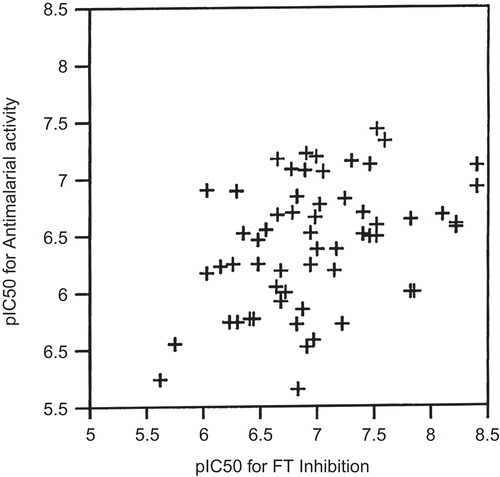
Along the same lines, we attempted to see if a correlation exists in the available large data set of reported benzophenones. We plotted the experimental pIC50 of both FT inhibition and antimalarial activities for 60 benzophenones, and found that the correlation between these two activities was greatly improved compared to the previous reports (). That, combined with the contour maps analysis, thus provides some evidence that the antimalarial activity of the benzophenones is based on the inhibition of farnesyltransferase. However, the considerable scatter in may be partly caused by differences between Pf FT and yeast FT.
Figure 4. Correlation of pIC50 against P. falciparum (antimalarial activity) to pIC50 against yeast farnesyltransferase (FT inhibition) for 60 of the 2,5-diaminobenzophenones considered in this work.
We conclude that the SAR trends with respect to the in vitro antimalarial parasite inhibitory activity in culture and the in vitro yeast FT inhibition assay exerted by the benzophenones are correlated to some extent. This is corroborated by the fact that we obtained almost the same property contours for two different activities for a large data set of benzophenones. The lack of strong correlation is due to limitations of the experiments, including different membrane-crossing properties of some of the compounds, and FT amino acid sequence differences between yeast and Pf species.
Compounds 4, 5, 18, 27, 35, 52, 78, 85, and 89 were outliers for both CoMFA and CoMSIA models. In a previous reportCitation22, the behavior of 89 was interpreted clearly by docking study using a homology model of yeast FT, which showed that the 2-naphthyl moiety was likely to occupy the aryl binding site better than other substituents. Compound 35 (which is an outlier in our QSAR modeling) is the only reported cinnamic acid amide benzophenone derivative containing a furylaryl moiety, and might have suffered from solubility troubles in the antimalarial assay medium, as predicted by the original authorsCitation35. The outlier behavior of the other compounds is hard to understand, especially for 4, 5, 18, and 27, even based on previous docking study of these compoundsCitation37; these four compounds fit into our previous antimalarial modelsCitation32 very well, so solubility issues are unlikely to be able to explain the outlier behavior. It is possible that the outliers result from structural differences between yeast FT and Pf FT or are just caused by experimental error; this could be determined through further study of farnesyltransferase and its inhibitors.
Conclusion
To summarize, as part of our continuing effort to find novel lead compounds for antimalarial drugs, we performed a 3D-QSAR investigation of farnesyltransferase inhibition activities, using the CoMFA and CoMSIA methods, for 95 compounds based on a 2,5-diaminobenzophenone scaffold selected from the work of Schlitzer et al. By thorough study, we found that steric, electrostatic, and hydrophobic features are key factors that impact the bioactivity, whereas the hydrogen bond donor and acceptor fields had no significant correlation with the bioactivity. For steric and electrostatic properties, CoMFA fields were far more interpretive than CoMSIA fields. We prepared a model which combined the steric and electrostatic fields of CoMFA with the hydrophobic field of CoMSIA; the model showed a significant improvement in prediction compared with the CoMFA-only steric and electrostatic field model. The steric, electrostatic, and hydrophobic fields were also the ones found to be important in our previous study on in vitro antimalarial activity of 92 2,5-diaminobenzophenones, including some of the same compounds used in this work, and the contour maps in this work were similar to those in the previous work, even though the models were for different experimental activities. Combining the results of the contour maps with correlation analysis of farnesyltransferase inhibition activities versus antimalarial activities for 60 out of the 95 compounds, we conclude that the yeast FT inhibition activity of the diaminobenzophenones is weakly correlated to their antimalarial activity, providing some support for the hypothesis that FT inhibition is their mechanism of action for killing P. falciparum.
Acknowledgements
Funding from the University of Mississippi, including from its Summer Research Institute for Undergraduates (for SRC) and Office of Research & Sponsored Programs; National Center for Zoonotic, Vector-borne, and Enteric Diseases (code CK) of the Centers for Disease Control and Prevention (CDC) (U01/CI000211); National Science Foundation (EPS-0556308); as well as the Laboratory for Applied Drug Design and Synthesis and MCSR computing facilities are greatly appreciated. SP is a Natural Products Neuroscience Fellow (P20 RR021929 from National Center for Research Resources (NCRR), National Institutes of Health (NIH)). This investigation was conducted in a facility constructed with support from research facilities improvement program C06 RR-14503-01 from the NIH National Center for Research Resources.
References
- Bell DR, Jorgensen P, Christophel EM, Palmer KL. Malaria risk estimation of the malaria burden. Nature 2005;437:E3–4.
- Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The global distribution of clinical episodes of plasmodium falciparum malaria. Nature 2005;434:214–17.
- Rosenthal PJ. Antimalarial drug discovery: old and new approaches. J Exp Biol 2003;206:3735–44.
- Schlitzer M. Malaria chemotherapeutics part 1: History of antimalarial drug development, currently used therapeutics, and drugs in clinical development. ChemMedChem 2007;2:944–86.
- Baird JK. Effectiveness of antimalarial drugs. N Engl J Med 2005;352:1565–77.
- Brady RL, Cameron A. Structure-based approaches to the development of novel anti-malarials. Curr Drug Target 2004;5:137–49.
- Eastman RT, White J, Hucke O, Bauer K, Yokoyama K, Nallan L, Chakrabarti D, Verlinde CLMJ, Gelb MH, Rathod PK, Van Voorhis WC. Resistance to a protein farnesyltransferase inhibitor in Plasmodium falciparum. J Biol Chem 2005;280:13554–9.
- Fidock DA, Rosenthal PJ, Croft SL, Brun R, Nwaka S. Antimalarial drug discovery: efficacy models for compound screening. Nat Rev Drug Discov 2004;3:509–20.
- Wiesner J, Ortmann R, Jomaa H, Schlitzer M. New antimalarial drugs. Angew Chem Int Ed Engl 2003;42:5274–93.
- Eastman RT, Buckner FS, Yokoyama K, Gelb MH, Van Voorhis WC. Thematic review series: lipid posttranslational modifications. Fighting parasitic disease by blocking protein farnesylation. J Lipid Res 2006;47:233–40.
- Gelb MH, Van Voorhis WC, Buckner FS, Yokoyama K, Eastman R, Carpenter EP, Panethymitaki C, Brown KA, Smith DF. Protein farnesyl and n-myristoyl transferases: piggy-back medicinal chemistry targets for the development of antitrypanosomatid and antimalarial therapeutics. Mol Biochem Parasitol 2003;126:155–63.
- Appels NMGM, Beijnen JH, Schellens JHM. Development of farnesyl transferase inhibitors: a review. Oncologist 2005;10:565–78.
- Dinsmore CJ, Bell IM. Inhibitors of farnesyltransferase and geranylgeranyltransferase-I for antitumor therapy: substrate-based design, conformational constraint and biological activity. Curr Top Med Chem 2003;3:1075–93.
- Pan J, Yeung S-CJ. Recent advances in understanding the antineoplastic mechanisms of farnesyltransferase inhibitors. Cancer Res 2005;65:9109–12.
- Sebti SM, Hamilton AD. Farnesyltransferase and geranylgeranyl transferase I inhibitors and cancer therapy: lessons from mechanism and bench-to-bedside translational studies. Oncogene 2000;19:6584–93.
- Carrico D, Ohkanda J, Kendrick H, Yokoyama K, Blaskovich MA, Bucher CJ, Buckner FS, Van Voorhis WC, Chakrabarti D, Croft SL, Gelb MH, Sebti SM, Hamilton AD. In vitro and in vivo antimalarial activity of peptidomimetic protein farnesyltransferase inhibitors with improved membrane permeability. Bioorg Med Chem 2004;12:6517–26.
- Nallan L, Bauer KD, Bendale P, Rivas K, Yokoyama K, Horney CP, Pendyala PR, Floyd D, Lombardo LJ, Williams DK, Hamilton A, Sebti S, Windsor WT, Weber PC, Buckner FS, Chakrabarti D, Gelb MH, VanVoorhis WC. Protein farnesyltransferase inhibitors exhibit potent antimalarial activity. J Med Chem 2005;48:3704–13.
- Wiesner J, Kettler K, Sakowski J, Ortmann R, Katzin AM, Kimura EA, Silber K, Klebe G, Jomaa H, Schlitzer M. Farnesyltransferase inhibitors inhibit the growth of malaria parasites in vitro and in vivo. Angew Chem Int Ed Engl 2004;43:251–4.
- Kettler K, Sakowski J, Silber K, Sattler I, Klebe G, Schlitzer M. Non-thiol farnesyltransferase inhibitors: N-(4-acylamino-3-benzoylphenyl)-3-[5-(4-nitrophenyl)-2-furyl]acrylic acid amides. Bioorg Med Chem 2003;11:1521–30.
- Kettler K, Wiesner J, Fucik K, Sakowski J, Ortmann R, Dahse H-M, Jomaa H, Schlitzer M. 2-(Aminoacylamino)benzophenones: farnesyltransferase inhibition and antimalarial activity. Pharmazie 2005;60:677–82.
- Kettler K, Wiesner J, Silber K, Haebel P, Ortmann R, Sattler I, Dahse H-M, Jomaa H, Klebe G, Schlitzer M. Non-thiol farnesyltransferase inhibitors: N-(4-aminoacylamino-3-benzoylphenyl)-3-[5-(4-nitrophenyl)-2 furyl]acrylic acid amides and their antimalarial activity. Eur J Med Chem 2005;40:93–101.
- Mitsch A, Bohm M, Wißner P, Sattler I, Schlitzer M. Non-thiol farnesyltransferase inhibitors: utilization of an aryl binding site by 5-arylacryloylaminobenzophenones. Bioorg Med Chem 2002;10:2657–62.
- Mitsch A, Wißner P, Silber K, Haebel P, Sattler I, Klebe G, Schlitzer M. Non-thiol farnesyltransferase inhibitors: N-(4-tolylacetylamino-3-benzoylphenyl)-3-arylfurylacrylic acid amides. Bioorg Med Chem 2004;12:4585–600.
- Sakowski J, Sattler I, Schlitzer M. Non-thiol farnesyltransferase inhibitors: N-(4-acylamino-3-benzoylphenyl)-4-nitrocinnamic acid amides. Bioorg Med Chem 2002;10:233–9.
- Cramer RD III, Patterson DE, Bunce JD. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc 1988;110:5959–67.
- Klebe G, Abraham U, Mietzner T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J Med Chem 1994;37:4130–46.
- Equbal T, Silakari O, Ravikumar M. Exploring three-dimensional quantitative structural activity relationship (3D-QSAR) analysis of SCH 66336 (Sarasar) analogues of farnesyltransferase inhibitors. Eur J Med Chem 2008;43:204–9.
- Murumkar PR, Giridhar R, Yadav MR. 3D-quantitative structure-activity relationship studies on benzothiadiazepine hydroxamates as inhibitors of tumor necrosis factor-alpha converting enzyme. Chem Biol Drug Des 2008;71:363–73.
- Puntambekar DS, Giridhar R, Yadav MR. Understanding the antitumor activity of novel tricyclicpiperazinyl derivatives as farnesyltransferase inhibitors using CoMFA and CoMSIA. Eur J Med Chem 2006;41:1279–92.
- Puntambekar DS, Giridhar R, Yadav MR. Insights into the structural requirements of farnesyltransferase inhibitors as potential anti-tumor agents based on 3D-QSAR CoMFA and CoMSIA models. Eur J Med Chem 2008;43:142–54.
- Sung ND, Cho YK, Kwon BM, Hyun KH, Kim CK. 3D QSAR studies on cinnamaldehyde analogues as farnesyl protein transferase inhibitors. Arch Pharm Res 2004;27:1001–8.
- Xie A, Sivaprakasam P, Doerksen RJ. 3D-QSAR analysis of antimalarial farnesyltransferase inhibitors based on a 2,5-diaminobenzophenone scaffold. Bioorg Med Chem 2006;14:7311–23.
- Sybyl 7.2. St. Louis, MO: Tripos, Inc., 2005.
- Grubbs F. Procedures for detecting outlying observations in samples. Technometrics 1969;11:1–21.
- Fucik K, Kettler K, Wiesner J, Ortmann R, Unterreitmeier D, Krauss J, Bracher F, Jomaa H, Schlitzer M. 2-(Arylpropionylamino)- and 2-(arylacryloylamino)benzophenones: farnesyltransferase inhibition and antimalarial activity. Pharmazie 2004;59:744–52.
- Wiesner J, Ortmann R, Mitsch A, Wißner P, Sattler I, Jomaa H, Schlitzer M. Inhibitors of farnesyltransferase: 5-arylacryloylaminobenzophenones show antimalarial activity. Pharmazie 2003;58:288–9.
- Bohm M, Mitsch A, Wißner P, Sattler I, Schlitzer M. Exploration of novel aryl binding sites of farnesyltransferase using molecular modeling and benzophenone-based farnesyltransferase inhibitors. J Med Chem 2001;44:3117–24.