Abstract
The activities of novel Cbz-N-protected α-aminophosphonic phenyl esters, analogs of leucine (1–15) and phenylalanine (17–29), which are substituted at the phenyl ester rings, as well as of their peptidic derivatives (31–43), were investigated for their inhibitory effects on chymotrypsin and subtilisin. The chemical nature and position of the examined substituents clearly demonstrated a strong structure–activity relationship. Among all synthesized compounds the most potent phosphonic-type inhibitors of subtilisin and chymotrypsin were identified, with k2/Ki values 114,380 M−1s−1 and 307,380 M−1s−1, respectively.
Introduction
Chymotrypsin-like serine proteases such as human mast cell chymase, cathepsin G, and microorganisms’ serine proteases of the subtilisin family (S8) are considered to be attractive targets for new inhibitor development and the design of potential new drugs. It was shown that inhibition of the subtilisin-like enzyme of the Cryptosporidium spp. parasite significantly diminished infection in cell culture, emphasizing the potential of this family of enzymes as a drug targetCitation1. A dual-active inhibitor of leukocyte protease cathepsin G and mast cell chymase has been proposed as a drug candidate for the simultaneous treatment of asthma and chronic obstructive pulmonary diseaseCitation2. One of the selective human mast cell chymase inhibitors was active as an anti-inflammatory agent in several animal models of inflammationCitation3. The inhibitors of mast cell chymase may also be useful for preventing cardiovascular disease, and many compounds, including phosphonate-type chymase inhibitor Suc-Val-Pro-PheP(OPh)2, have proven their activity in several animal modelsCitation4.
α-Aminoalkylphosphonate diphenyl esters and their peptidyl derivatives are potent and selective irreversible inhibitors of different classes of serine proteasesCitation5–Citation7. They represent a delicate chemical balance between phosphorus atom electrophilicity and specificity of action. The phosphonate phosphorus atom, in contrast to the routinely used diagnostic agent for the detection of serine protease activity diisopropylphosphofluoridate (DIPF), does not have strong electrophilic properties. Additionally, the peptide fragment of peptidyl α-aminoalkylphosphonate derivatives provides specific interactions with the enzyme, which allows the nucleophilic hydroxyl group of Ser195 located in the active site to attack the moderately electrophilic phosphorus atom of the inhibitor. The stability of the resulting enzyme–phosphonic inhibitor complexes range from 8 h for chymotrypsin to more than 3 days for trypsin and human neutrophil elastaseCitation7.
Reported α-aminoalkylphosphonic inhibitors of chymotrypsin and chymotrypsin-related enzymes such as cathepsin G and rat mast cell protease II contain unsubstituted diphenyl ester ringsCitation6. Only one publication describes a series of halophenyl diester derivatives of α-aminophosphonic inhibitors of chymotrypsin, which showed higher activity than their unsubstituted counterpart structuresCitation8.
It is worth mentioning that subtilisin and chymotrypsin are not evolutionarily relatedCitation9. The three-dimensional structures of the molecules are not similar, and the catalytic triad composed of His/Asp/Ser in chymotrypsin occurs in a different sequence in subtilisin, Asp/His/Ser. It is of interest to compare the inhibition of both enzymes by the phosphonate-type inhibitors, and in this report we describe a series of new phosphonic analogs of phenylalanine and leucine as chymotrypsin and subtilisin inhibitors.
Materials and methods
Chemistry
All reagents used in the experiments were purchased from Merck, Sigma-Aldrich, Lancaster, and Fluka. Melting points were determined on a Boëtius, Nagema Rapido, PHMK 05 apparatus. 1H and 31P nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AC-TM DRX 300 (300.13 MHz (1H), 121.51 MHz (31P)) spectrometer.
General procedure for synthesis of Cbz-N-protected α-aminoalkylphosphonate diphenyl esters
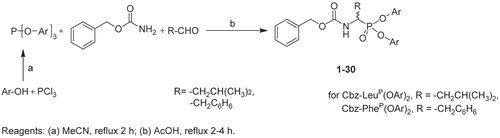
N-benzyloxycarbonyl diphenyl esters of α-aminoalkylphosphonates were obtained as racemic mixtures by the α-amidoalkylation reaction of triphenyl phosphiteCitation6. The triphenyl phosphites were prepared by heating phosphorus trichloride (1 eq) with corresponding phenol (3 eq) at 80°C in acetonitrile for 2 h (). The volatile products were evaporated under reduced pressure and the obtained crude phosphite was used directly in the α-amidoalkylation reaction without purification; to the equimolar amount of phosphite, isovaleric aldehyde, or phenylacetaldehyde, benzyl carbamate and acetic acid were added and the mixture was refluxed for 2–4 h. Reaction progress was monitored by thin layer chromatography (TLC). After the reaction mixture was evaporated, the resulting oil was dissolved in methanol and left for crystallization at –20°C. The obtained product was filtered, washed with cold methanol, and recrystallized if necessary.
Scheme 1. Synthesis of Cbz-N-protected derivatives of α-aminoalkylphosphonate diphenyl esters. Reagents: (a) MeCN, reflux 2 h; (b) AcOH, reflux 2–4 h.
General procedure for synthesis of dipeptides with C-terminal α-aminoalkylphosphonate diphenyl esters
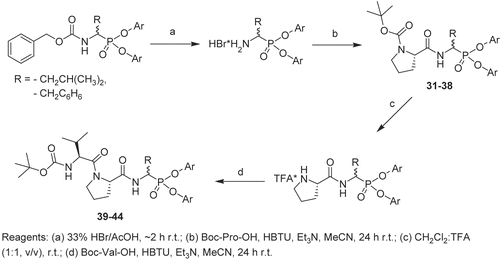
For synthesis of the dipeptide derivatives of α-aminoalkylphosphonate diphenyl esters, hydrobromides of α-aminoalkylphosphonate diphenyl esters were used (): the racemic mixture of Cbz-N-protected α-aminoalkylphosphonate diphenyl ester was dissolved in 33% HBr/AcOH solution and allowed to react for 2 h at room temperature; the reaction mixture was evaporated under reduced pressure, and the resulting oil was dissolved in diethyl ether and left at –20°C for crystallization; the product was then filtered, washed with cold diethyl ether, and dried in air. Boc-l-Pro-OH (1.1 eq), triethylamine (2.5 eq), and HBTU (O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate) (1.2 eq) were added successively to a solution of the α-aminoalkylphosphonate diphenyl ester hydrobromide (1 eq) in acetonitrile. The reaction was performed at room temperature for 24 h. The solution was diluted with ethyl acetate, washed with brine, 5% KHSO4, brine, 5% NaHCO3, and finally with water. The organic layer was dried over MgSO4, filtered, and evaporated. All dipeptides containing C-terminal α-aminoalkylphosphonate diphenyl esters were further purified by column chromatography on silica gel.
Scheme 2. Synthesis of peptidyl derivatives of α-aminoalkylphosphonate diphenyl esters. Reagents: (a) 33% HBr/AcOH, ~2 h room temperature (r.t.); (b) Boc-Pro-OH, HBTU, Et3N, MeCN, 24 h r.t.; (c) CH2Cl2/TFA (1:1, v/v), r.t.; (d) Boc-Val-OH, HBTU, Et3N, MeCN, 24 h r.t.
General procedure for synthesis of tripeptides containing C-terminal α-aminoalkylphosphonate diphenyl esters
The crude Boc-protected dipeptide containing an α-aminoalkylphosphonate diphenyl ester at the C-terminus was dissolved in a dichloromethane/trifluoroacetic acid (TFA) (1:1, v/v) cleavage mixture, and the progress of the reaction was followed by TLC. When the starting material was not present the mixture was evaporated, re-dissolved in dry toluene, and evaporated under reduced pressure. The resulting oil was dissolved in acetonitrile, and coupling with Boc-l-Val-OH was performed by the method described above (). The final compounds were purified by column chromatography.
Scheme 3. Kinetic scheme for serine protease inhibition by diphenyl α-aminophosphonate derivatives.
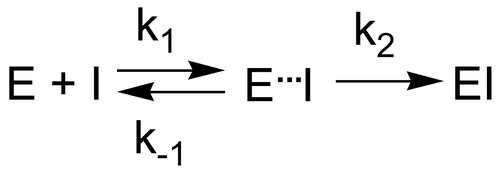
Procedure for synthesis of Suc-Val-Pro-PheP(OPh)2
Compound 42 was used for the synthesis of Suc-Val-Pro-PheP(OPh)2. Boc-Val-Pro-PheP(OPh)2 (1 eq) was dissolved in CH2Cl2/TFA (1:1, v/v) solution; the mixture was then evaporated, and after dissolving it in ethyl acetate, succinic anhydride (1 eq) and triethylamine (1.1 eq) were added. After 1 h a second equivalent of triethylamine was added. The reaction was performed at room temperature for 2 h and volatile matter was removed under reduced pressure. The obtained residue was diluted with ethyl acetate, and washed with 5% citric acid solution and water. The organic layer was dried over MgSO4, filtered, and evaporated, yielding the desired compound.
Compound 1
Yield 38%; m.p. 126°C; 31P NMR (CDCl3): 20.37 (s); 1H NMR (CDCl3): 0.98 (d, J = 5.79 Hz, 6H, 2 × CH3), 1.61–2.16 (m, 3H, CH2, CH), 2.47 (s, 6H, 2 × CH3), 4.61 (m, 1H, CHP), 5.18 (d, J = 10.37 Hz, 1H, NH), 5.06 (m, 2H, ArCH2O), 6.91–7.30 (m, 13H, Ar-H).
Compound 2
Yield 68%; m.p. 161°C; 31P NMR (CDCl3): 19.03 (s); 1H NMR (CDCl3): 0.89 (d, J =5 .87 Hz, 6H, 2 × CH3), 1.19 (s, 18H, 6 × CH3), 1.63–2.09 (m, 3H, CH2, CH), 4.48 (m, 1H, CHP), 5.05 (m, 2H, ArCH2O), 6.66–7.27 (m, 13H, Ar-H).
Compound 3
Yield 22%; m.p. 91–105°C; 31P NMR (CDCl3): 18.18 (s); 1H NMR (CDCl3): 0.99 (d, J = 5.52 Hz, 6H, 2 × CH3), 1.73–1.84 (m, 3H, CH2, CH), 2.00–2.32 (m, 6 × CH3), 4.60 (m, 1H, CHP), 5.08 (m, 2H, ArCH2O), 6.76–7.34 (m, 9H, Ar-H).
Compound 4
Yield 51%; m.p. 111°C; 31P NMR (CDCl3): 18.75 (s); 1H NMR (CDCl3): 0.98 (d, J = 5.44 Hz, 6H, 2 × CH3), 1.73–1.84 (m, 3H, CH2, CH), 2.41 (d, J = 13.92 Hz, 18H, 6 × CH3), 4.58 (m, 1H, CHP), 5.11 (d, J = 10.53 Hz, 1H, NH), 5.03 (m, 2H, ArCH2O), 6.50–7.37 (m, 9H, Ar-H).
Compound 5
Yield 48%; m.p. 103°C; 31P NMR (CDCl3): 19.03 (s); 1H NMR (CDCl3): 0.98 (d, J = 5.8 Hz, 6H, 2 × CH3), 1.22 (m, 12H, 4 × CH3), 1.73–1.82 (m, 3H, CH2, CH), 2.83 (m, 2H, 2 × CH), 4.60 (s, 1H, CHP), 5.15 (s, 1H, NH), 5.17 (m, 1H, ArCH2O), 6.97–7.35 (m, 13H, Ar-H).
Compound 6
Yield 47%; m.p. 104°C; 31P NMR (CDCl3): 18.84 (s); 1H NMR (CDCl3): 0.98 (d, J = 5.64 Hz, 6H, 2 × CH3), 1.87 (m, 3H, CH2, CH), 2.29 (m, 12H, 4 × CH3), 4.61 (m, 1H, CHP), 5.13 (m, 2H, ArCH2O), 6.82–7.33 (m, 11H, Ar-H).
Compound 7
Yield 29%; m.p. 115°C; 31P NMR (CDCl3): 18.40 (s); 1H NMR (CDCl3): 1.01 (d, J = 5.04 Hz, 6H, 2 × CH3), 1.91 (m, 3H, CH2, CH), 2.25 (m, 12H, 4 × CH3), 4.68 (m, 1H, CHP), 5.20 (m, 2H, ArCH2O), 6.82–7.43 (m, 11H, Ar-H).
Compound 8
Yield 43%; m.p. 87°C; 31P NMR (CDCl3): 19.53 (s); 1H NMR (CDCl3): 0.90 (d, J = 5.68 Hz, 6H, 2 × CH3), 1.79 (m, 3H, CH2, CH), 3.82 (s, 6H, 2 × COOCH3), 4.52 (m, 1H, CHP), 5.01–5.16 (m, 3H, ArCH2O, NH), 6.92–7.93 (m, 13H, Ar-H).
Compound 9
Yield 38%; m.p. 118°C; 31P NMR (CDCl3): 18.82 (s); 1H NMR (CDCl3): (d, J = 5.86 Hz, 6H, 2 × CH3), 1.77 (m, 3H, CH2, CH), 2.31 (d, J = 7.10 Hz, 6H, 2 × CH3), 4.61 (m, 1H, CHP), 5.11 (s, 1H, NH), 5.14 (m, 1H, 2H, ArCH2O), 6.96–7.35 (m, 13H, Ar-H).
Compound 10
Yield 63%; m.p. 107°C; 31P NMR (CDCl3): 18.43 (s); 1H NMR (CDCl3): 0.99 (m, 6H, 2 × CH3), 1.81 (m, 3H, CH2, CH), 2.25 (m, 12H, 4 × CH3), 4.68 (m, 1H, CHP), 4.98–5.16 (m, 3H, ArCH2O, NH), 6.90–7.30 (m, 11H, Ar-H).
Compound 11
Yield 62%; m.p. 116°C; 31P NMR (CDCl3): 18.64 (s); 1H NMR (CDCl3): 1.00 (m, 6H, 2 × CH3), 1.78 (m, 3H, CH2, CH), 2.25 (m, 6H, 2 × CH3), 4.66 (m, 1H, CHP), 4.99–5.16 (m, 3H, ArCH2O, NH), 6.96–7.35 (m, 13H, Ar-H).
Compound 12
Yield 45%; m.p. 86°C; 31P NMR (CDCl3): 18.99 (s); 1H NMR (CDCl3): 0.98 (d, J = 5.60 Hz, 6H, 2 × CH3), 1.22 (m, 6H, 2 × CH3), 1.72 (m, 3H, CH2, CH), 2.55 (m, 4H, 2 × CH2), 4.60 (m, 1H, CHP), 5.15 (s, 1H, NH), 5.17 (m, 1H, ArCH2O), 7.27–7.32 (m, 13H, Ar-H).
Compound 13
Yield 45%; m.p. 142°C; 31P NMR (CDCl3): 19.43 (s); 1H NMR (CDCl3): 0.98 (d, J = 5.96 Hz, 6H, 2 × CH3), 1.88 (m, 3H, CH2, CH), 4.63 (m, 1H, CHP), 5.19 (s, 1H, NH), 5.11 (m, 2H, ArCH2O), 6.90–7.35 (m, 13H, Ar-H).
Compound 14
Yield 61%; m.p. 59–60°C; 31P NMR (CDCl3): 19.29 (s); 1H NMR (CDCl3): 0.99 (d, J = 5.98 Hz, 6H, 2 × CH3), 1.86 (m, 3H, CH2, CH), 4.65 (m, 1H, CHP), 4.96–5.18 (m, 3H, ArCH2O, NH), 6.80–7.36 (m, 13H, Ar-H).
Compound 15
Yield 53%; m.p. 59°C; 31P NMR (CDCl3): 18.76 (s); 1H NMR (CDCl3): 0.98 (d, J = 5.68 Hz, 6H, 2 × CH3), 1.69–2.30 (m, 3H, CH2, CH), 3.80 (d, J = 11.17 Hz, 6H, 2 × CH3), 4.65 (m, 1H, CHP), 5.13 (m, 2H, ArCH2O), 5.19 (s, 1H, NH), 6.40–7.33 (m, 13H, Ar-H).
Compound 16
Yield 37%; m.p. 112°C; 31P NMR (CDCl3): 17.97 (s); 1H NMR (CDCl3): 0.98 (d, J = 5.60 Hz, 6H, 2 × CH3), 1.93 (m, 3H, CH2, CH), 4.65 (m, 1H, CHP), 5.00 (s, 1H, ArCH2O), 5.11 (d, J = 8.99 Hz, 1H, NH), 7.13–7.32 (m, 15H, Ar-H).
Compound 17
Yield 38%; m.p. 137°C; 31P NMR (CDCl3): 19.51 (s); 1H NMR (CDCl3): 3.09 (d, J = 12.32 Hz, 6H, 2 × CH3), 3.4 ( m, 2H, CH2), 4.8 (m, 1H, CHP), 4.95 (s, 2H, ArCH2O), 5.05 (d, J = 9.96 Hz, 1H, NH), 7.19–7.80 (m, 18H, Ar-H).
Compound 18
Yield 47%; m.p. 112°C; 31P NMR (CDCl3): 17.58 (s); 1H NMR (CDCl3): 1.18 (m, 6H, 2 × CH3), 2.52 (m, 4H, 2 × CH2,), 3.30 (m, 2H, CH2), 4.7 (m, 1H, CHP), 4.92 (s, 2H, ArCH2O), 5.14 (d, J = 10.2 Hz, 1H, NH), 6.91–7.23 (m, 18H, Ar-H).
Compound 19
Yield 38%; m.p. 119°C; 31P NMR (CDCl3): 17.28 (s); 1H NMR (CDCl3): 3.16 (m, 6H, 2 × CH3), 3.85 (m, 2H, CH2), 5.70 (m, 1H, CHP), 5.8 (m, 2H, ArCH2O), 6.07 (d, J = 10.43 Hz, 1H, NH), 7.8–8.14 (m, 18H, Ar-H).
Compound 20
Yield 88%; m.p. 99–102°C; 31P NMR (CDCl3): 18.11 (s); 1H NMR (CDCl3): 3.40 (m, 2H, CH2,), 3.69 (d, J = 8.24 Hz, 6H, 2vCH3), 4.70 (m, 1H, CHP), 4.92 (s, 2H, ArCH2O), 5.12 (d, J = 10.57 Hz, 1H, NH), 6.65–7.23 (m, 18H, Ar-H).
Compound 21
Yield 28%; m.p. 129°C; 31P NMR (CDCl3): 17.63 (s); 1H NMR (CDCl3): 1.16 (t, J = 8.12 Hz, 12H, 4 × CH3), 2.94 (m, 2H, 2 × CH), 3.37 (m, 2H, CH2), 4.77 (m, 1H, CHP), 4.81 (s, 2H, ArCH2O), 5.36 (d, J = 9.95 Hz, 1H, NH), 6.93–7.25 (m, 18H, Ar-H).
Compound 22
Yield 54%; m.p. 99°C; 31P NMR (CDCl3): 18.04 (s); 1H NMR (CDCl3): 2.90–3.00 (m, 2H, CH2), 4.73 (m, 1H, CHP), 4.92 (m, 2H, ArCH2O), 5.09 (d, J = 10.50 Hz, 1H, NH), 7.00–7.23 (m, 18H, Ar-H).
Compound 23
Yield 40%; m.p. 103°C; 31P NMR (CDCl3): 17.08 (s); 1H NMR (CDCl3): 2.17 (m, 12H, 4 × CH3), 2.9–3.4 (m, 2H, CH2), 4.8 (m, 1H, CHP), 4.90 (m, 2H, ArCH2O), 5.15 (d, J = 10.42 Hz, 1H, NH), 6.86–7.28 (m, 16H, Ar-H).
Compound 24
Yield 22%; m.p. 139°C; 31P NMR (CDCl3): 17.64 (s); 1H NMR (CDCl3): 1.09 (m, 18H, 6 × CH3), 3.18 (m, 2H, CH2), 4.78 (m, 1H, CHP), 4.9 (s, 2H, ArCH2O), 5.02 (d, J = 10.0, 1H, NH), 6.71–7.13 (m, 18H, Ar-H).
Compound 25
Yield 10%; m.p. 151–152.5°C; 31P NMR (CDCl3): 17.64 (s); 1H NMR (CDCl3): 2.98–3.42 (m, 2H, CH2), 3.87 (s, 6H, 2 × CH3), 4.83 (m, 1H, CHP), 4.97 (s, 2H, ArCH2O), 5.12 (d, J = 10.5 Hz, 1H, NH), 6.7–7.13 (m, 18H, Ar-H).
Compound 26
Yield 40%; m.p. 97–99°C; 31P NMR (CDCl3): 18.98 (s); 1H NMR (CDCl3): 2.40 (d, J = 6.52 Hz, 6H, 2 × CH3), 2.90–3.36 (m, 2H, CH2), 4.70 (m, 1H, CHP), 4.93 (s, 2H, ArCH2O), 5.07 (d, J = 10.9 Hz, 1H, NH), 6.69–7.28 (m, 18H, Ar-H).
Compound 27
Yield 41%; m.p. 72°C; 31P NMR (CDCl3): 17.45 (s); 1H NMR (CDCl3): 2.2 (m, 12H, 4 × CH3), 2.9–3.3 (m, 2H, CH2), 4.91 (m, 1H, CHP), 5.30 (d, J = 10.42 Hz, 1H, NH), 5.02 (m, 2H, ArCH2O), 6.54–7.23 (m, 16H, Ar-H).
Compound 28
Yield 50%; m.p. 101–104°C; 31P NMR (CDCl3): 17.91 (s); 1H NMR (CDCl3): 2.90–3.02 (m, 2H, CH2), 4.75 (m, 1H, CHP), 4.91 (m, 2H, ArCH2O), 5.31 (d, J = 10.30 Hz, 1H, NH), 7.00–7.23 (m, 18H, Ar-H).
Compound 29
Yield 36%; m.p. 155°C; 31P NMR (CDCl3): 17.36 (s); 1H NMR (CDCl3): 2.19 (m, 18H, 6 × CH3), 2.90–3.38 (m, 2H, CH2), 4.72 (m, 1H, CHP), 4.88(m, 2H, ArCH2O), 5.1 (d, J = 10.08 Hz, 1H, NH), 6.66–7.23 (m, 14H, Ar-H).
Compound 30
Yield 48%; m.p. 124.5–125.5°C; 31P NMR (CDCl3): 18.46 (s); 1H NMR (CDCl3): 2.98–3.48 (m, 2H, CH2), 4.82 (m, 1H, CHP), 4.88 (s, 2H, ArCH2O), 5.15 (d, J = 10.17 Hz, 1H, NH), 7.06–7.29 (m, 20H, Ar-H).
Compound 31
Yield 43%; m.p. 54°C; 31P NMR (CDCl3): 19.62 (s); 1H NMR (CDCl3): 0.81 (m, 6H, 2 × CH3), 1.31 (s, 9H, 3 × CH3), 1.66–2.21 (m, 7H, 3 × CH2, CH), 3.26 (m, 2H, CH2N), 3.76 (s, 6H, 2 × CH3), 4.17 (m, 1H, CHCO), 4.78 (m, 1H, CHP), 7.27–7.75 (m, 8H, Ar-H).
Compound 32
Yield 66%; m.p. 53–55°C; 31P NMR (CDCl3): 18.76 (s), 18.58 (s); 1H NMR (CDCl3): 0.88 (m, 6H, 2 × CH3), 1.34 (s, 9H, 3 × CH3), 1.62–1.98 (m, 7H, 3 × CH2, CH), 2.53 (m, 18H, 6 × CH3), 3.4 (m, 2H, CH2N), 4.2 (m, 1H, CHCO), 4.8 (m, 1H, CHP), 6.67–7.31 (m, 4H, Ar-H).
Compound 33
Yield 18%; m.p. 53°C; 31P NMR (CDCl3): 18.78 (s), 18.61 (s); 1H NMR (CDCl3): 0.88 (m, 6H, 2 × CH3), 1.36 (s, 9H, 3 × CH3), 1.62–2.02 (m, 5H, 2 × CH2, CH), 2.07–2.09 (m, 6H, 2 × CH3), 2.12–2.21 (m, 2H, CH2), 3.27 (m, 2H, CH2N), 4.14 (m, 1H, CHCO), 4.86 (m, 1H, CHP), 6.97–7.28 (m, 4H, Ar-H).
Compound 34
Yield 30%; m.p. 49–51°C; 31P NMR (CDCl3): 18.86 (s); 1H NMR (CDCl3): 0.86 (d, J = 9.07 Hz, 6H, 2 × CH3), 1.37 (s, 9H, 3 × CH3), 1.63–2.02 (m, 5H, 2 × CH2, CH), 2.12 (m, 12H, 4 × CH3), 2.23 (m, 2H, CH2), 3.32 (m, 2H, CH2N), 4.20 (m, 1H, CHCO), 4.76 (m, 1H, CHP), 6.78–7.19 (m, 6H, Ar-H).
Compound 35
Yield 65%; m.p. 39.5–40.5°C; 31P NMR (CDCl3): 18.78 (s), 18.79 (s); 1H NMR (CDCl3): 0.93 (m, 6H, 2 × CH3), 1.44 (s, 9H, 3×CH3), 1.69–2.04 (m, 7H, 3 × CH2, CH), 2.15–2.30 (m, 6H, 2 × CH3), 3.40 (m, 2H, CH2N), 4.24 (d, J = 16.22 Hz, 1H, CHCO), 4.85 (m, 1H, CHP), 7.00–7.28 (m, 8H, Ar-H).
Compound 36
Yield 90%; m.p. 96°C; 31P NMR (CDCl3): 19.00 (s), 18.83 (s); 1H NMR (CDCl3): 0.85 (m, 6H, 2 × CH3), 1.37 (s, 9H, 3 × CH3), 1.65–1.90 (m, 7H, 3 × CH2, CH), 3.50 (m, 2H, CH2N), 3.6 (t, J = 2.32 Hz, 6H, 2 × CH3), 4.30 (m, 1H, CHCO), 5.2 (m, 1H, CHP), 6.72–7.19 (m, 8H, Ar-H).
Compound 37
Yield 19%; m.p. 69°C; 31P NMR (CDCl3): 17.83 (s), 17.63 (s); 1H NMR (CDCl3): 2.33 (m, 9H, 3 × CH3), 1.36–1.98 (m, 4H, 2 × CH2), 3.41 (m, 4H, CH2N, CH2), 4.19 (m, 1H, CHCO), 5.18 (m, 1H, CHP), 7.04–7.67 (m, 15H, Ar-H).
Compound 38
Yield 90%; m.p. 49–52°C; 31P NMR (CDCl3): 18.24 (s), 18.16 (s); 1H NMR (CDCl3): 2.5 (s, 9H, 3 × CH3), 1.65–1.90 (m, 4H, 2 × CH2), 2.35 (s, 6H, 2 × CH3), 3.40 (m, 4H, CH2N, CH2), 4.12 (m, 1H, CHCO), 5.00 (m, 1H, CHP), 5.19 (s, 1H, NH), 6.94–7.18 (m, 13H, Ar-H).
Compound 39
Yield 43%; m.p. 59°C;31P NMR (CDCl3): 18.51 (s), 18.63 (s); 1H NMR (CDCl3): 0.84 (m, 12H, 4 × CH3), 1.34 (s, 9H, 3 × CH3), 1.65–1.86 (m, 8H, 3 × CH2, 2 × CH), 2.13–2.16 (m, 18H, 6 × CH3), 3.50 (m, 3H, CH2N), 4.20 (m, 1H, CH), 4.36 (m, 1H, CHCO), 4.7 (m, 1H, CHP), 5.2 (d, J = 10.52 Hz, 1H, NH), 6.70–7.0 (m, 4H, Ar-H).
Compound 40
Yield 23%; m.p. 62°C; 31P NMR (CDCl3): 19.83 (s), 19.55(s); 1H NMR (CDCl3): 0.92 (m, 12H, 4 × CH3), 1.36 (s, 9H, 3 × CH3), 1.60–2.10 (m, 8H, 3 × CH2, 2 × CH), 3.62–3.68 (m, 2H, CH2N), 3.69 (s, 6H, 2 × CH3), 4.22 (m, 1H, CH), 4.44 (m, 1H, CHCO), 4.59 (m, 1H, CHP), 5.28 (d, J = 9.28 Hz, 1H, NH), 6.70–7.19 (m, 4H, Ar-H).
Compound 41
Yield 32%; m.p. 52–58°C; 31P NMR (CDCl3): 18.69 (s), 18.92 (s); 1H NMR (CDCl3): 0.92 (m, 12H, 4 × CH3), 1.22 (m, 18H, 6 × CH3), 1.36 (s, 9H, 3 × CH3), 1.67–1.88 (m, 8H, 3 × CH2, 2 × CH), 3.50–3.67 (m, 2H, CH2N), 4.12 (m, 1H, CH), 4.60 (m, 1H, CHCO), 4.75 (m, 1H, CHP), 5.10 (d, J = 8.9 Hz, 1H, NH), 5.24 (d, J = 11.32 Hz, 1H, NH), 6.98–7.24 (m, 4H, Ar-H).
Compound 42
Yield 23%; m.p. 68°C; 31P NMR (CDCl3): 17.35 (s), 17.37 (s); 1H NMR (CDCl3): 0.68 (m, 6H, 2 × CH3), 1.20 (s, 9H, 3 × CH3), 1.50–1.79 (m, 5H, 2 × CH2, CH), 3.14–3.39 (m, 2H, CH2N), 3.4 (m, 2H, CH2), 3.99 (m, 1H, CH), 4.3 (m, 1H, CHCO), 5.12 (m, 1H, CHP), 5.16 (m, 1H, NH), 6.87–7.07 (m. 3H, Ar-H).
Compound 43
Yield 80%; m.p. 72–76°C; 31P NMR (CDCl3): 18.30 (s), 18.26 (s); 1H NMR (CDCl3): 0.88 (m, 6H, 2 × CH3), 1.37 (s, 9H, 3 × CH3), 1.65–1.80 (m, 5H, 2 × CH2, CH), 2.35 (m, 6H, 2 × CH3), 3.15 (m, 2H, CH2N), 3.5 (m, 2H, CH2), 4.99 (m, 1H, NH), 4.16 (m, 1H, CHCO), 5.11 (m, 1H, CHP), 5.16 (m, 1H, NH), 6.79–7.01 (m, 8H, Ar-H).
Suc-Val-Pro-PheP(OPh)2
Yield 70%; 31P NMR (CDCl3): 18.48 (s); 1H NMR (CDCl3): 1.80 (m, 6H, 2 × CH3), 1.83–1.96 (m, 5H, 2 × CH2, CH), 2.54 (m, 4H, 2 × CH2), 3.20 (m, 2H, CH2N), 3.5 (m, 2H, CHCO), 4.40 (m, 1H, CH), 5.11 (m, 1H, CHP), 7.07–7.15 (m, 17H, Ar-H, 2 × NH), 7.79 (s, 1H, OH).
Enzyme inhibition assay
Bovine chymotrypsin and subtilisin A from Bacillus licheniformis were purchased from Calbiochem and Sigma-Aldrich, respectively. The substrate used for both enzymes, Suc-Ala-Ala-Pro-Phe-AMC, was purchased from Calbiochem. The assay buffers were as follows: 100 mM HEPES, 500 mM NaCl, pH 7.5 containing 9% DMSO (dimethyl sulfoxide) for chymotrypsin; 50 mM Tris, 1M NaCl, pH 7.5 containing 0.01% Triton X-100 for subtilisin. The intensity of the fluorescence was measured using a Molecular Devices Gemini XPS Microplate Spectrofluorometer (ex 350 nm, em 460 nm). Measured Km (Michaelis constant) values were 60 μM (for subtilisin) and 70 μM (for chymotrypsin). The Ki (inhibition constant) values were calculated from the inhibition of AMC (7-amido-4-methyl coumarin) formation by different concentrations of the tested compound. The hyperbolic model of inhibition was used to calculate Ki and k2 valuesCitation10–Citation12. The inhibitory activity of synthesized compounds was determined by the progress curve method under pseudo-first-order conditions ([I]0>>[E]0, and with less than 5% substrate conversion). Control curves in the absence of inhibitor were linear. The rate of substrate hydrolysis was continuously monitored by measuring the rate of increase of fluorescence at 460 nm (λexc 350 nm). The pseudo-first-order rate constants (kobs) for the inhibition of chymotrypsin and subtilisin as a function of time were determined according to the equation: AMCt = ν0[1 – exp(–kobst)]/kobs + AMC0, where AMCt is the fluorescence intensity at time t, AMC0 is the fluorescence intensity at time zero, and ν0 is the reaction velocity at time zero. By fitting the AMC vs. t data to this equation using nonlinear regression analysis the kobs values were obtained. The second-order rate constant (k2/Ki) was calculated from the slope of the linear part of the plot using the following equation: kobs/[I] = k2/(Ki + [I]). The obtained Ki and k2/Ki values of chymotrypsin and subtilisin inactivation by phosphonic analogs of leucine and phenylalanine and their di- and tripeptide derivatives are summarized in .
Results and discussion
Twenty eight novel, substituted at diphenyl ester rings Cbz-N-protected α-aminophosphonic analogs of leucine (1–15) and phenylalanine (17–29) as well as their peptidic derivatives (31–43) were synthesized by a previously described methodCitation6. Cbz-N-protected α-aminophosphonic analogs of leucine (16) and phenylalanine (30) unsubstituted at their diphenyl ester rings were included as reference compounds. The synthetic approach is presented in and starts with the synthesis of triphenyl phosphites.
An α-amidoalkylation reaction of triphenyl phosphites gives the desired Cbz-N-protected α-aminophosphonates as the racemic mixture, with a yield ranging from 10 to 68%. For the synthesis of their peptidyl derivatives, a standard HBTU coupling method was applied using hydrobromide salts of parent Cbz-deprotected α-aminoalkylphosphonate diphenyl esters ().
Inhibition of serine proteases by diphenyl α-aminophosphonate derivatives is a two step process (). The first step leads to the formation of a reversible noncovalent complex (Ki). The next step involves a first order reaction resulting in the formation of a covalent phosphonylated enzyme (k2).
The overall inactivation rate constant (k2/Ki) was measured by the progress curve method. We have set up an arbitrary activity criteria utilizing inhibitors with Ki > 20 μM and k2/Ki < 50 M−1s−1 as inactive compounds. Inactivation rate constants obtained for subtilisin and chymotrypsin by Cbz-LeuP(OAr)2 are shown in . Among all Cbz-LeuP(OAr)2 derivatives tested against subtilisin, only the structure bearing 4-t-butylphenyl diesters (2) showed moderate activity (k2/Ki = 253 M−1s−1). In contrast, chymotrypsin was inhibited by several phosphonic leucine analogs with compound 8, a 4-carboxymethylphenyl ester derivative displaying the highest inhibition rate constant value of the group at 61,770 M−1s−1.
Table 1. Ki, k2/Ki for inactivation of chymotrypsin and subtilisin by leucine related phosphonates.
The high inhibitory potency of compound 8 is the result of increased electrophilicity of the phosphorus atom caused by strong electrowithdrawing properties of the 4-carboxymethyl group. However, we do not consider these types of compounds to have practical value due to their increased sensitivity to hydrolysis. In contrast, derivatives 1 (4-mercaptomethylphenyl esters) and 15 (3-methoxyphenyl esters) do not carry an electrowithdrawing moiety and their high activity is most likely a result of additional interactions within the enzyme leaving the group binding side (S1′, S2′ pockets). Compounds 1 and 15 are the most potent single α-aminophosphonic leucine analog inhibitors of chymotrypsin reported so far, with k2/Ki values of 18,300 M−1s−1 and 8760 M−1s−1, respectively.
The activities of various diphenyl esters of phenylalanine analogs are presented in . For subtilisin the 4-carboxymethylphenyl (25) and 4-mercaptomethylphenyl (26) esters were found to be the most potent inactivators; however, their activity was not better than the unsubstituted diphenyl ester derivative 30. It seems that the substitution at the phenyl diester ring does not improve activity against subtilisin. In other words, the leaving group side of subtilisin is rather small, and does not accommodate the phenyl rings of any of the tested substituents. Interestingly, no such restriction was noticed for leucine analog 2. Derivative 17 with a 4-methylsulfonyl substitution had the highest potency against subtilisin with the k2/Ki value 47,100 M−1s−1; however, the presence of a strongly electron-accepting moiety in the para position on the phenyl phosphonate ester makes this derivative hydrolytically unstable and without practical value.
Table 2. Ki, k2/Ki for inactivation of chymotrypsin and subtilisin by phenylalanine related phosphonates.
Compounds 17 and 25 are also the most potent inhibitors of chymotrypsin within phosphonic analogs of phenylalanine, with k2/Ki values of 307,380 M−1s−1 and 300,000 M−1s−1 respectively. However, for reasons described above they do not possess practical application potential.
Dipeptide derivatives of α-aminoalkylphosphonate diphenyl esters with a Cbz-N-protecting group are the only ones reported in the literature. In general, such compounds display poor inhibitory potency against serine proteases compared even to single diphenyl phosphonic amino acid analogsCitation6,Citation7. Surprisingly, the Boc-protected dipeptidyl derivatives show quite good activity, especially against subtilisin (demonstrated in and 4).
Table 3. Ki, k2/Ki for inactivation of chymotrypsin and subtilisin by dipeptidyl derivatives of leucine related phosphonates.
Table 4. Ki, k2/Ki for inactivation of chymotrypsin and subtilisin by dipeptidyl derivatives of phenylalanine related phosphonates.
For example, Boc-Pro-LeuP(OAr)2, derivative 36 with a 4-methoxy substitution at the phenyl ester ring, displayed Ki = 7.05 μM and k2/Ki = 1930 M−1s−1 against subtilisin. The 4-mercaptomethyl derivative 38 is a good peptidic inhibitor of subtilisin, with Ki = 5.3 μM and k2/Ki = 1560 M−1s−1, and it also inactivates chymotrypsin with Ki = 0.73 μM and k2/Ki = 1600 M−1s−1.
The inhibition rate constants measured for tripeptide derivatives 39–43 are presented in and . Tripeptides containing leucine-related phosphonates are relatively weak chymotrypsin inhibitors, but derivative 40 (Ki = 0.06 μM and k2/Ki = 13,760 M−1s−1) is one of the most active subtilisin inhibitors found in this study. It is worth noting that no activity was found for tripeptide 41 with a 4-t-butylphenyl substitution. The corresponding single α-aminophosphonic acid derivative 2 was the only one active toward subtilisin in series 1–15. It could be speculated that the observed significant difference in activity resides in the mode of compound binding. The relatively outstanding activity of 2 could be the result of binding the 4-t-butylphenyl ester moiety in the S1 pocket of the enzyme in addition to “normal binding” (where the 2-methylpropyl constituent is bound to the S1 pocket). In the case of tripeptide derivative 41 with extended binding through the S3–S1 pockets this additional mode of binding is not possible.
Table 5. Ki, k2/Ki for inactivation of chymotrypsin and subtilisin by tripeptidyl derivatives of leucine related phosphonates.
Table 6. Ki, k2/Ki for inactivation of chymotrypsin and subtilisin by tripeptidyl derivatives of phenylalanine related phosphonates.
The activity of tripeptides with a C-terminal phosphonic analog of phenylalanine, 42 and 43, is similar to that of the most potent known phosphonate chymotrypsin inhibitor Suc-Val-Pro-PheP(OPh)2Citation6,Citation7. However, compound 43 with 4-mercaptomethylphenyl ester rings is the most active subtilisin inhibitor found in this study, with Ki= 0.093 μM and k2/Ki = 114,380 M−1s−1.
In conclusion, we have investigated the influence of substitutions at diphenyl ester rings in a series of α-aminoalkylphosphonates and their peptidyl derivatives on the inhibition of subtilisin and chymotrypsin. Our study clearly demonstrated a strong structure–activity relationship, and a new, highly potent phosphonate-type inhibitor of subtilisin, 43, was identified. For chymotrypsin, it seems, the substitution of the phosphonate diphenyl ester rings by a variety of different substituents at different positions does not significantly improve inhibitory activity.
Acknowledgments
The authors would like to thank Dr. Keri Smith for critical reading of the manuscript. This work was supported by the Wrocław University of Technology, grant no. 343622.
Declaration of interest: The authors report no conflicts of interest.
References
- Feng X, Akiyoshi DE, Widmer G, Tzipori S. Characterization of subtilase protease in Cryptosporidium parvum and C.hominis. J Parasitol 2007;93:619–26.
- De Garavilla L, Greco MN, Sukumar N, Chen ZW, Pineda AO, Mathews FS, Di Cera E, Giardino EC, Wells GI, Haertlein BJ, Kauffman JA, Corcoran TW, Derian CK, Eckardt AJ, Damiano BP, Andrade-Gordon P, Maryanoff BE. A novel, potent dual inhibitor of the leukocyte proteases cathepsin G and chymase: molecular mechanisms and anti-inflammatory activity in vivo. J Biol Chem 2005;280:18001–7.
- Greco MN, Hawkins MJ, Powell ET, Almond HR Jr, de Garavilla L, Hall J, Minor LK, Wang Y, Corcoran TW, Di Cera E, Cantwell AM, Savvides SN, Damiano BP, Maryanoff BE. Discovery of potent, selective, orally active, nonpeptide inhibitors of human mast cell chymase. J Med Chem 2007;50:1727–30.
- Takai S, Jin D, Muramatsu M, Okamoto Y, Miyazaki M. Therapeutic applications of chymase inhibitors in cardiovascular diseases and fibrosis. Eur J Pharmacol 2004;501:1–8.
- Oleksyszyn J, Powers JC. Irreversible inhibition of serine proteases by peptidyl derivatives of alpha-aminoalkylphosphonate diphenyl esters. Biochem Biophys Res Commun 1989;161:143–9.
- Oleksyszyn J, Powers JC. Irreversible inhibition of serine proteases by peptide derivatives of (alpha-aminoalkyl)phosphonate diphenyl esters. Biochemistry 1991;30:485–93.
- Oleksyszyn J, Powers JC. Amino acid and peptide phosphonate derivatives as specific inhibitors of serine peptidases. Methods Enzymol 1994;244:423–41.
- Boduszek B, Brown AD, Powers JC. Alpha-aminoalkylphosphonate di(chlorophenyl) esters as inhibitors of serine proteases. J Enzyme Inhib 1994;8:147–58.
- Rawlings ND, Barrett AJ. Families of serine proteases. Methods Enzymol 1994;244:19–61.
- Salvesen GS, Nagase H. Inhibition of proteolytic enzymes. In: Proteolytic Enzymes: A Practical Approach. Oxford: Oxford University Press, 1989:83–104.
- Knight CG. The characterization of enzyme inhibition. In: Proteinase Inhibitors. Amsterdam: Elsevier, 1986:23–51.
- Joossens J, Ali OM, El-Sayed I, Surpateanu G, Van der Veken P, Lambeir AM, Setyono-Han B, Foekens JA, Schneider A, Schmalix W, Haemers A, Augustyns K. Small, potent, and selective diaryl phosphonate inhibitors for urokinase-type plasminogen activator with in vivo antimetastatic properties. J Med Chem 2007;50:6638–46.