Abstract
The crucial role of the microtubule in cell division has identified tubulin as a target for the development of therapeutics for cancer; in particular, tubulin is a target for antineoplastic agents that act by interfering with the dynamic stability of microtubules. A molecular modeling study was carried out to accurately represent the complex structure and the binding mode of a new class of stilbene-based tubulin inhibitors that bind at the αβ-tubulin colchicine site. Computational docking along with HINT (Hydropathic INTeractions) score analysis fitted these inhibitors into the colchicine site and revealed detailed structure–activity information useful for inhibitor design. Quantitative analysis of the results was in good agreement with the in vitro antiproliferative activity of these derivatives (ranging from 3 nM to 100 μM) such that calculated and measured free energies of binding correlate with an r2 of 0.89 (standard error ± 0.85 kcal mol−1). This correlation suggests that the activity of unknown compounds may be predicted.
Introduction
Cancer is the leading cause of death in the USA and many developed countries. According to the World Health Organization Fact Sheet, 7.4 million people died of cancer worldwide in 2004, and more than 70% of those deaths were reported in low- and middle-income countries. The number of global cancer deaths is projected to increase 45% from 2007 to 2030Citation1,Citation2. Several different approaches have been implemented for prevention, early detection, diagnosis, and treatment of various types of cancer. In terms of treatment, there are a number of effective chemotherapeutic drugs in the market, with diverse mechanisms of action that target various stages of cancerous cells, with the main objective of stopping cell proliferation. Compounds that inhibit cell proliferation and exert cytotoxic activity by perturbing microtubule dynamics have been explored with some success.
Combretastatins, natural products derived from the bark of the South African tree Combretum caffrum, have been demonstrated to bind at the colchicine binding site and destabilize microtubule assembly and prevent spindle formation in mitotic cellsCitation3. The relatively simple structure and high affinity of combretastatins for the colchicine binding site has led to the synthesis and subsequent evaluation of a large number of analogs; novel compounds derived from this core continue to hold interest as potential therapeuticsCitation3–5. Stilbene and its analogs are structurally similar to combretastatin and are also able to bind to microtubules, suppress microtubule dynamics, and arrest the cell cycle at the G2/M phase that is associated with the induction of apoptosisCitation6,Citation7.
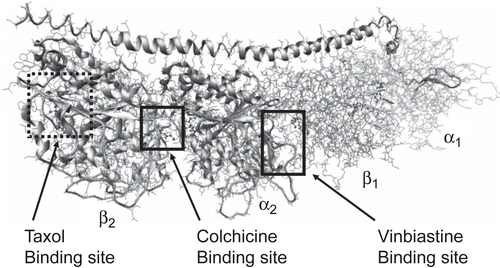
Microtubules are fundamental and essential structures constituting the cytoskeleton of eukaryotic cells; they also participate in the formation and stabilization of the mitotic spindle, and are thus critical in cell division. The basic building block of the microtubule is the tubulin protein. In 1998, Nogales et al. reported the first three-dimensional structural model of tubulinCitation8. This model shows that the tubulin protein exists as a dimer consisting of two monomeric units that are almost identical in structure. Each monomer is formed by a core of two β sheets surrounded by α helices, with each binding a guanine nucleotide. In addition to the nucleotide binding site, αβ-tubulin has three distinct binding sites that have been investigated for ligands in the vinca alkaloid, colchicinoid, and taxol classes ()Citation9. There are several drugs from these classes that bind to tubulin and disrupt microtubule dynamicsCitation10. Numerous synthetic and semi-synthetic analogs have been prepared and evaluated, but all have issues that make the development of additional classes of drugs that destabilize microtubules and inhibit cell proliferation desirable for treating drug-resistant cancers. As the synthesis of stilbene analogs is relatively facile, our goal for this study was to create a quantitative structure–activity model of these compounds that would guide the synthesis of new, potentially more efficacious stilbene derivatives. Thus, we report here on the structural requirements for interaction between stilbene analogs and tubulin using computational docking and hydropathic scoring to fit multiple stilbene analogs into the colchicine binding site of tubulin.
Figure 1. The tubulin-colchicine:RB3-SLD complex. The complex includes alternating tubulin αβ heterodimers, with the colchicine binding site at the intradimer interface, the taxol binding site on the β subunit, and the vinblastine binding site at the interdimer interface of the αβ subunit.
Materials and methods
Synthesis
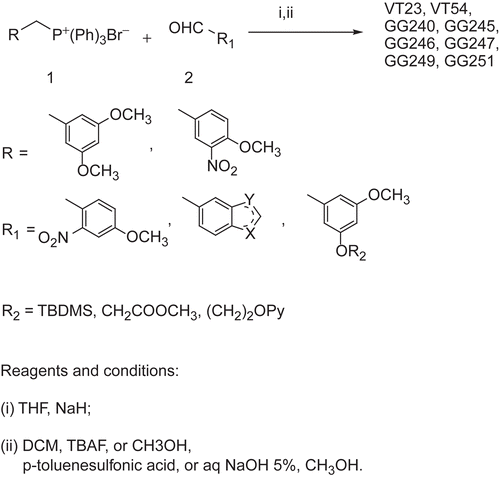
Synthesis of stilbenes 5c and 6c was performed as described by Roberti et al.Citation11. Stilbenes VT23, VT54, GG240, GG245, GG246, GG247, GG249, and GG251 were easily prepared as described in and previously reported. Briefly, a Wittig reaction between the available phosphonium bromide 1 and the appropriate aldehyde 2 was performed in tetrahydrofuran (THF) using sodium hydride as the base, to give a mixture of E and Z stereoisomers that were, in turn, separated by flash chromatography. The nitro derivatives were easily converted into amino moieties with zinc in glacial acetic acid solution. The terbutyldimethylsilyl (TBDMS) ether protecting group was removed with tetrabutylammonium fluoride (TBAF). Alkaline hydrolysis of the methyl ester gives the desired carboxylic acid derivative.
Scheme 1. Compound synthesis.
The 2-(3,4,5-trimethoxybenzoyl) benzo[b]furan derivatives CTR103, CTR106, and CTR105 were synthesized by condensing salicylaldehyde and its 5-methoxy and 5-nitro derivatives, respectively, with 2-bromo-39,49,59- trimethoxyacetophenone and potassium carbonate in refluxing acetone (). The benzo[b]thiophene analog CTR104 was obtained by condensation of 2-mercaptobenzaldehyde and 2-bromo-39,49,59-trimethoxyacetophenone in refluxing acetone in the presence of potassium carbonate as base ().
Scheme 2. Synthesis of CTR103–CTR106.
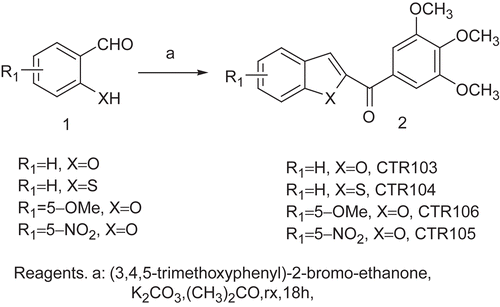
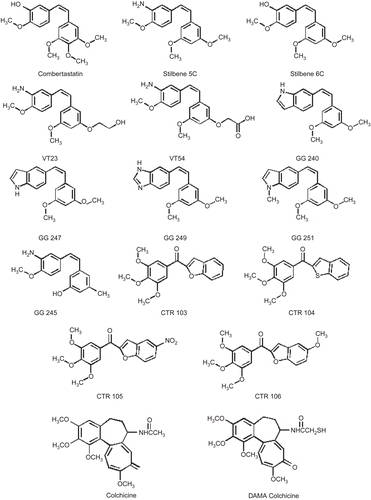
Figure 2. Stilbene derivatives.
Antiproliferative activity of stilbenes against human tumor cell lines
The antiproliferative activity of stilbene derivatives was determined by Alamar Blue™ staining. In brief, cells were grown in 96-well plates and treated with 0, 0.01, 0.03, 0.1, 0.3, 1.0, and 3.0 μM of the stilbene for 48 h before being harvested for Alamar Blue™ staining. In this staining, 1/10 volume of Alamar Blue™ solution was added to each well, and optical density (OD) at 570 and 600 nm was determined by a microplate reader. The percentage of growth inhibition was calculated according to the manufacturer’s formula as follows: [(117,216 × A570) – (80,586 × A600)]/[(117,216 × A°570) – (80,586 × A°600)] × 100. In this formula, A570 is the absorbance of the treated samples at 570 nm; A600 is the absorbance of the treated samples at 600 nm; A°570 is the absorbance of the untreated samples at 570 nm; and A°600 is the absorbance of the untreated samples at 600 nm. The two constants, 117,216 and 80,586, are the extinction coefficients of Alamar Blue™ at 570 and 600 nm respectively. Each concentration was repeated in triplicate.
Model building
The X-ray crystal structure (3.58 å) of αβ-tubulin complexed with DAMA-colchicineCitation12 (Protein Data Bank (PDB) code: 1SA0) was used in this study. The stathmin-like domain and the C and D subunits were removed from the model. After hydrogen atoms were added to the model, their positions were optimized to an energy gradient of 0.005 kcal-å/mol with the Tripos force field (in Sybyl 7.3 software) while keeping heavy atom positions fixed. The models for stilbene analogs were constructed using Sybyl 7.3 (www.tripos.com) and optimized similarly.
Docking
Computational docking was carried out using the genetic algorithm-based ligand docking program GOLD 3.0Citation13. GOLD explores ligand conformations fairly exhaustively, and also provides limited flexibility to protein side chains with hydroxyl groups by reorienting the hydrogen bond donor and acceptor groups. The active site was defined by using colchicine as the reference molecule in the protein active site, creating an approximate radius of 10 å around the reference molecule using the GOLD cavity detection algorithm. Because of the relatively poor resolution of the X-ray crystal structure and following the approach of Nguyen et al.Citation14, GOLD docking was carried out with template similarity constraint. This constraint biases the conformation of docked ligands toward a given solution. The trimethoxyphenyl fragment of colchicine was used as the template for biasing the pose of all ligands. In this study we performed 100 GOLD genetic algorithm runs as opposed to the default of 10, and early termination of ligand docking was switched off. All other parameters were as the defaults. To evaluate and validate GOLD performance, the co-crystallized ligand DAMA-colchicineCitation12 was extracted and docked. GOLD accurately reproduced the experimentally observed binding mode of DAMA-colchicine in αβ-tubulin. Combretastatin was docked first, and the resulting model was scored and optimized. The remaining stilbene analogs were docked and optimized using combretastatin as a reference within 0.5 å root mean square distance (RMSD) by employing the same parameters. Docked ligands were scored using the HINT (Hydropathic INTeractions) force field scoring function (see below) and iteratively optimized for maximal interaction.
Dockings with different/optional constraints such as enforced hydrogen bonds, hydrophobic regions, and scaffold match were also explored. For hydrogen bond constraints, docking was biased so that the ligands made hydrogen bonds with Asn258, Ser178, Asn101, and the backbone amides of Ala180 and Val181. For regional hydrophobic constraints, the ligand positions were constrained by defining a hydrophobic sphere where the trimethoxyphenyl moiety of colchicines was positioned. Then specific ligand atoms to be docked in the hydrophobic region of the active site were defined. Alternatively, scaffold match constraints were used to place the ligand at a specific position within the active site. Generally, however, because the active site is rather featureless, constraint or template-free docking was not successful.
Hydropathic scoring
The HINT scoring functionCitation15 (version 3.11S β) was used to investigate the structural aspects of the interactions by analyzing and ranking the GOLD docking solutions. HINT evaluates and scores each atom–atom interaction in a biomolecular complex using a parameter set derived from solvation partition coefficients for 1-octanol/water. LogPo/w is a thermodynamic parameter that can be directly correlated with free energyCitation16. The HINT model describes specific interactions between two molecules as:
where a is the hydrophobic atom constant derived from LogPo/w, S is the solvent accessible surface area, T is a function that differentiates polar–polar interactions (acid–acid, acid–base, or base–base), and R, r are functions of the distance between atoms i and j as previously describedCitation17. The binding score, bij, describes the specific atom–atom interaction between atoms i and j, whereas B describes the total interaction. For selection of the optimum docked conformation and to further differentiate the relative binding efficacy of the stilbene ligands, interaction scores were calculated for each pose found by docking. The protein and ligands were partitioned as distinct molecules. “Essential” hydrogen atoms, i.e. only those attached to polar atoms (N, O, S, P), were explicitly considered in the model and assigned HINT constants. The inferred solvent model, where each residue is partitioned based on its hydrogen count, was applied. The solvent accessible surface area for the amide nitrogens of the protein backbone were corrected with the “+ 20” option. Finally, HINT scores were plotted against experimental binding free energy.
Results and discussion
Antiproliferative activity of stilbene analogs
The biological activity of all compounds was tested in UCI-101 ovarian cancer cells; qualitatively similar trends were observed in MDA-MB231 breast cancer cellsCitation18. Compounds could be separated into three groups according to their potency. Group A contains the most potent compounds, including combretastatin, stilbene 5C, GG251, colchicine, DAMA-colchicine, VT23, and stilbene 6C, with IC50 less than 100 nM. Group B contains GG240, GG247, GG245, and GG249, with IC50 in the intermediate range between 0.3 and 1.0 μM. Group C is not active, with IC50 above 5 μM ().
Table 1. Experimental IC50 and docking results for stilbene and colchicine derivatives.
The colchicine binding site
A number of groups have performed modeling studies on the colchicine binding site because it is recognized as a potential target for anticancer drug developmentCitation14. However, the low resolution of the available crystal structures for tubulin have made it difficult to fully delineate the essential structural and functional features involved in tubulin inhibition. In a low-resolution crystal structure model, our knowledge of the correct orientation of side chains is limited by ambiguities in position and orientation, because the experimental electron density envelopes are generally featureless. The crystal structure of tubulin protein used in this study has a fairly poor resolution of 3.58 å, which somewhat undermines our ability to (structure-based) design highly selective ligandsCitation12. In earlier studies of the colchicine binding site, we carried out computational docking studies along with hydropathic interaction analysis on a family of substituted pyrroles and were able to represent the complex structure and the binding modes of the pyrrole class of inhibitorsCitation19.
We use the HINT modelCitation15 as our scoring tool for docking, and to quantitatively and qualitatively evaluate hydropathic interactions. This tool effectively identifies critical binding interactions. The HINT program, which has been invented and developed in our laboratory, calculates empirical atom-based hydropathic parameters derived from the experimental data from solvent partitioning, i.e. the partition coefficient (LogPo/w). LogPo/w is a thermodynamic parameter representing the free energy of solvent transfer between the solvents (1-octanol and water), which encodes all significant intermolecular and intramolecular non-covalent interactions implicated in drug binding. The algorithm evaluates and scores each atom–atom interaction in a biomolecular complex, and this can be directly correlated with the free energy of bindingCitation13.
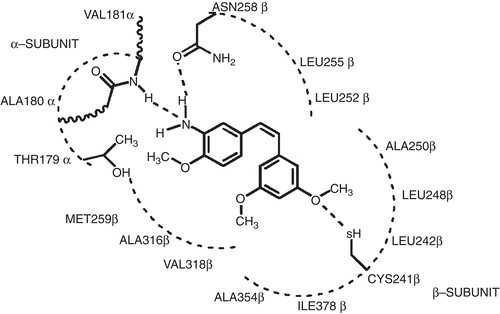
Binding modes for each stilbene analog were investigated to understand the steric, electrostatic, and hydropathic features of the colchicine binding site. A molecular modeling study of docking these ligands into the colchicine site of αβ-tubulin was carried out in order to accurately represent the complex structure. The colchicine binding site is positioned at the interface between the α and β subunits of the tubulin protein, with the major part of it buried in the β subunit and lined by helices 7 and 8. The cavity, which is funnel shaped, has a volume of about 600 å, and opens up toward the α subunit of the interface, surrounded by residues Asn101α, Thr179α, Ala180α, Val181α, Thr314β, Asn349β, Asn350β, and Lys352β. The other, β subunit, end of the cavity is surrounded by residues Tyr202β, Val238β, Thr239β, Cys241β, Leu242β, Leu248β, Leu252β, Leu255β, Ile378β, and Val318β, and forms the narrow, funnel end-like part of the cavity. The predominance of hydrophobic residues confers a strong hydrophobic character to this part of the cavity. At the wider portion, the cavity is surrounded by Ala250β, Asp251β, Lys254β, Asn258β, Met259β, Ala316β, Ala317β, Thr353β, and Ala354β, making it moderately polar/moderately hydrophobic. DAMA-colchicine (and presumably colchicine) is snugly positioned in the crystal structure of the complex. The trimethoxyphenyl (TMP) moiety of colchicine is positioned in the pocket such that it sits snugly in the narrow hydrophobic region of the pocket with one of its methoxy oxygens involved in hydrogen bonding with the thiol of Cys241β. Colchicine also forms hydrogen bonds with the backbone amides of Ala180α and Val181α.
The structural complex as reported in the X-ray structure was refined through ligand functional group and protein side chain optimization, as incorporated in the HINT program. Iteratively, colchicine was translated and rotated and optimized for interactions. Taking colchicine as the template, combretastatin was computationally docked and scored. Similar to above, the combretastatin–tubulin complex was optimized. Next, the stilbene analogs were docked, this time taking combretastatin as the template, followed by HINT functional group and protein side chain optimization. The docked models of the stilbene analogs fit within the pharmacophore model proposed by Nguyen et al.Citation14, and are similar to the models we reported earlier for the pyrrole derivatives bound to αβ-tubulinCitation19. These studies, coupled with HINT interaction analyses, are able to describe the complex structure and the binding modes of stilbene inhibitors. Note that HINT scores are very sensitive, and slight positional differences are detectable in the scores. Qualitative analyses of the results showed general agreement with the experimental in vitro antiproliferative activity for these derivatives.
Structure–activity binding relationships
Structural analysis of the binding pocket identified important intermolecular interactions that mediate binding. The stilbene analogs were clustered into three activity sets in order to study them in detail (see ). The first (A) comprised compounds that showed activity from 3 nM to 60 nM IC50. The second (B) consisted of ligands with IC50 values ranging from 0.3 to 1 μM. The remaining ligands, with IC50 values of 5 μM and above, constituted the third set (C). HINT analysis reveals significant detail concerning the forces orienting these ligands in the binding site. First, hydrophobic interactions are the dominating force contributing toward the stability of the complexes, with additional hydrogen bonding interactions anchoring the ligands in the cavity. The TMP moieties of colchicine and combretastatin are positioned within the narrow hydrophobic region of the cavity, while the carbonyl oxygen on the unsaturated seven-member ring of colchicine and the hydroxyl group on the combretastatin B ring are anchored through strong hydrogen bonding interactions with Asn258β and the Ala180α and Val181α backbone amides.
First, in examining the docked models for activity subset A (), it is interesting to note that, although the stilbene compounds and combretastatin are quite dissimilar structurally to colchicine (excepting the TMP moiety), they are generally positioned in the binding pocket with essentially the same mode. In the case of stilbene 5C and GG251, the hydrophobic substituted phenyl ring fits snugly in the hydrophobic (narrow funnel) region of the binding pocket that superimposes on the TMP moiety of colchicines (). In fact, stilbene 5C and GG251 have a very similar binding mode to that of combretastatin, the major contribution toward binding coming from hydrophobic interactions. The methoxy substituted phenyls are positioned deep in the hydrophobic cavity surrounded by Cys241β, Leu242β, Leu248β, Ala250β, Leu255β, Ala354β, and Ile378β, all of which contribute to favorable hydrophobic–hydrophobic binding. The phenyl ring of stilbene 5c and GG251 fits in a hydrophobic glove formed by Leu248β and Leu255β. Favorable polar interaction with Asn101α, Cys241β, and Asn258β also contributes to the tight binding. The NH2 group on the B ring of stilbene 5C faces toward the polar opening, and is stabilized with a strong hydrogen bond to the amide oxygen of Asn258β, with a length of 2.55 å. Another set of strong hydrogen bonds is formed between the amide backbone of Val181α and the amine on the B ring of stilbene 5C with a hydrogen bond distance of 3.384 å. However, in the case of GG251, the hydrogen bonding interaction is not observed with the Asn258β and Val181α amide backbone; instead, the complex is predominantly stabilized through hydrophobic interactions. Similar interactions are observed with stilbene 6C and VT23, with some subtle differences. In the case of stilbene 6C, although the hydroxyl group is retained on the B ring as in combretastatin, the removal of the methoxy group from position 4 of the TMP moiety results in loss of activity. However, this loss is offset by introducing the NH2 group on ring B in stilbene 5C, thus accounting for the slight differences in their activity. In the case of VT23, the extension of the methoxy chain on the TMP moiety results in further loss of activity, probably due to geometric constraints enforced by the narrow hydrophobic region of the cavity.
Figure 3. Stilbene analogs docked at the colchicine binding site on αβ-tubulin. (a) Substituted stilbenes with activity of sub-μM IC50. (b) Compounds with IC50 ranging from 0.3 to 1.0 μM. (c) Compounds with IC50 values above 5 μM.
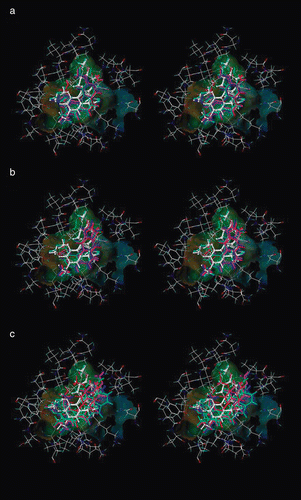
Figure 4. Representation of interactions of stilbene 5C in the colchicine active site of the tubulin protein.
On analyzing subset B (compounds GG240, GG247, GG245, and GG249) (), docked ligands in the low μM range, it can be seen that these ligands are relatively similar to the subset A ligands. In this set of compounds, substitution on ring B, in the cases of GG240, GG247, and GG249 the indole-carrying ring and in GG245 the amine- and methoxy-carrying ring, is varied. These substitutions result in a 10-fold decrease in activity in compounds GG240 and GG247, where the N-methyl substitution is removed from the indole ring of GG251. Flipping the ring, as in the 6- and 5-substituted indole ring of GG240 and GG247, does not have any significant affect on activity. Activity is further decreased if the indole ring is replaced by a benzimidazole ring as in compound GG249, and a similar loss in activity is noted in GG245 where the methoxy group of the stilbene 5C methoxyphenyl moiety is replaced by a methyl group and a hydroxyl group, confirming the importance of the methoxyphenyl moiety on ring A in binding. The benzofuran and benzothiophene analogs in the CTR series of compounds (subset C) () are similar to 2-aroylindoles, where the 2-aroylindole ring is replaced by benzofuran and benzothiophene. Our docked complexes with the CTR series of compounds agree with the Nguyen et al. pharmacophore modelCitation14, but the rings are inappropriately substituted to make the required contacts with the binding site residues—leading to poor activity of these compounds.
Predictive models for ligand binding
presents the correlation between the experimental binding (ΔGbinding as calculated from IC50) in kcal mol−1 and HINT scores for the synthetic stilbene analogs in this study. The trend represented by this plot indicates that higher scoring complexes are generally among those with more favorable free energies of binding, while lower scoring complexes are generally those with unfavorable binding. The correlation equation:
has an r2 = 0.8993 and a standard error of ±0.85 kcal mol−1.
Figure 5. Correlation plot between free energy of binding ΔG vs. HINT score. The line represents the regression for ΔG vs. HINT score for all protein–ligand complexes in this study.
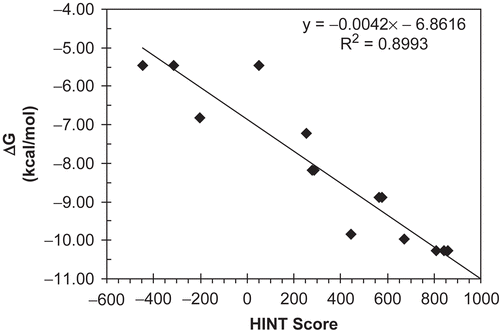
The IC50 values, antiproliferative activities of the compounds, are being taken in this work as approximations of binding affinity. The scoring function does not take into account cell permeability, and completely ignores whether or not the compound could in vivo or in vitro be accessible to the binding site. However, the similar structures of these compounds and the fairly narrow range of LogP suggest that these properties would be similar, if not the same, for all of these compounds, and thus can be ignored while comparing the compounds within the series. We believe that the model of is predictive such that it can distinguish the active (subset A) ligands from the inactive (subset B) ligands with reasonable confidence. Refinement of the model with additional data will further improve the understanding of the binding process and predictive ability of the model.
Conclusions
The aim of this study was to accurately represent the complex structures and the binding mode of a new class of stilbene based tubulin inhibitors. Both qualitative and quantitative analysis of the results suggested that the model was in general agreement with the in vitro antiproliferative activity observed experimentally for these compounds. A good correlation between the modeled interaction energies and estimated free energies of binding calculated from IC50 values suggest that our model is able to represent the complex structures and binding modes of inhibitors, and under some circumstances be predictive with respect to new members of the stilbene series. We believe that we can identify active ligands from inactive ligands with reasonable confidence; our analysis has provided a rationale for selecting substituents that will yield more tightly binding analogs.
Acknowledgements
Declaration of interest: The authors report no conflicts of interest.
References
- World Health Organization. Fact Sheet (Cancer) No. 297 Available at http://www.who.int/mediacentre/factsheets/fs297/en/index.html.
- American Cancer Society. Global Cancer Facts and Figures 2007. Available at http://www.cancer.org
- Tron GC, Pirali T, Sorba G, Pagliai F, Busacca S, Genazzani AA. Medicinal chemistry of combretastatin A4: present and future directions. J Med Chem 2006;49:3033–44.
- Bellina F, Cauteruccio S, Monti S, Rossi R. Novel imidazole-based combretastatin A-4 analogues: evaluation of their in vitro antitumor activity and molecular modeling study of their binding to the colchicine site of tubulin. Bioorg Med Chem Lett 2006;16:5757–62.
- Ji Y, Tian R, Lin W. QSAR and molecular docking study of a series of combretastatin analogues tubulin inhibitors. LNCS 2007;4689:436.
- Belleri M, Ribatti D, Nicoli S, Cotelli F, Forti L, Vannini V, Stivala LA, Presta M. Antiangiogenic and vascular-targeting activity of the microtubule-destabilizing trans-resveratrol derivative 3,5,49-trimethoxystilbene. Mol Pharmacol 2005;67:1451–9.
- Cao TM, Durrant D, Tripathi A, Liu J, Tsai S, Kellogg GE, Simoni D, Lee RM. Stilbene derivatives that are colchicine site microtubule inhibitors have anti-leukemic activity and minimal systemic toxicity. Am J Hematol 2008;83:390–7.
- Nogales E, Wolf SG, Downing KH. Structure of the αβ tubulin dimer by electron crystallography. Nature 1998;391:199–203.
- Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev 2004;4:253–65.
- Jordan A, Hadfield JA, Lawrence NJ, McGown AT. Tubulin as a target for anticancer drugs: agents which interact with the mitotic spindle. Med Res Rev 1998;18:259–96.
- Roberti M, Pizzirani D, Simoni D, Rondanin R, Baruchello R, Bonora C, Buscemi F, Grimaudo S, Tolomeo M. Synthesis and biological evaluation of resveratrol and analogues as apoptosis-inducing agents. J Med Chem 2003;46:3546–54.
- Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Insight into tubulin regulation from a complex with colchicine and a stathmin like domain. Nature 2004;428:198–202.
- Jones G, Willett P, Glen RC, Leach AR, Taylor RJ. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997;267:727–48.
- Nguyen TL, McGrath C, Hermone AR, Burnett JC, Zaharevitz DW, Day BW, Wipf P, Hamel E, Gussio R. A common pharmacophore for a diverse set of colchicine site inhibitors using a structure-based approach. J Med Chem 2005;48:6107–16.
- Kellogg GE, Abraham DJ. Hydrophobicity: is LogP(o/w) more than the sum of its parts? Eur J Med Chem 2000;35:651–61.
- Cozzini P, Fornabaio M, Marabotti A, Abraham DJ, Kellogg GE, Mozzarelli A. Simple, intuitive calculations of free energy of binding for protein-ligand complexes. 1. Models without explicit constrained water. J Med Chem 2002;45:2469–83.
- Spyrakis F, Amadasi A, Fornabaio, M, Abraham DJ, Mozzarelli A, Kellogg GE, Cozzini P. The consequences of scoring docked ligand conformations using free energy correlations. Eur J Med Chem 2007;42:921–33.
- Durrant DE, Richards J, Tripathi A, Kellogg GE, Marchetti P, Eleopra M, Grisolia G, Simoni D, Lee RM. Development of water soluble derivatives of cis-3,49,5-trimethoxy-39-aminostilbene for optimization and use in cancer therapy. Invest New Drugs 2009;27:41–52.
- Tripathi A, Fornabio M, Kellogg GE, Gupton JT, Gewirtz DA, Mooberry SL. Docking and hydropathic scoring of polysubstituted pyrroles compounds with anti-tubulin activity. Bioorg Med Chem 2008;16:2235–42.