Abstract
In this article the synthesis of new 1H-(2′-pyridyl)-3-methyl-5-hydroxypyrazole and 1H-(2′-pyridyl)-3-phenyl-5-hydroxypyrazole complexes with palladium(II) ions is reported. The structures of obtained compounds have been characterized by X-ray crystallography and DFT (density functional theory) calculations. The cytotoxicity of complexes and ligands has been examined for two human leukemia cell lines (HL-60 and NALM-6) and one human melanoma cell line (WM-115). The palladium(II) complex with 1H-(2′-pyridyl)-3-phenyl-5-hydroxypyrazole has been shown to possess greater activity than carboplatin against the WM-115 melanoma cell line. Additionally, the ligands’ tautomeric forms existence in different solvents (chloroform, methanol, DMSO) has been characterized by 1H nuclear magnetic resonance (NMR) analysis and DFT calculations. The obtained results have been compared with those from other studies of similar compounds.
Introduction
The development of antitumor metal complexes began with the first reports of Rosenberg and co-workers that some platinum complexes possess the ability to inhibit the growth of cancer cellsCitation1,Citation2. The most active compound was found to be cis-diamminedichloroplatinum(II): cisplatin. It has found application in the treatment of many types of cancer, including testicular, ovarian, head and neck, bladder, and non-small cell lung cancer. The main problem of cisplatin treatment is its toxicity. Cisplatin is nephrotoxic, neurotoxic, and ototoxic. It also possesses strong emetogenic properties. However, this is not the only problem with cisplatin usage. Some types of cancer exhibit intrinsic or acquired resistance to this drug. Cisplatin also has to be administered intravenously. All these problems stimulated research for new metal complexes with anticancer activityCitation3–6. Besides the new platinum complexes, the search for better anticancer agents was conducted among complexes of many other metal ions, such as gold(I) and gold(III)Citation7, ruthenium(II) and ruthenium(III)Citation8,Citation9, copper(II)Citation10–12, and palladium(II)Citation13,Citation14 ions. In particular, the palladium ion seems to be interesting, as it exhibits a similar coordination mode to that of the Pt(II) ion, and its complexes also possess square-planar geometryCitation15. Additionally, differences in reactivity of Pd(II) and Pt(II) complexesCitation16 may provoke greater activity of one of them. During the development of new antitumor compounds among metal complexes, many different ligands were examined, for example aliphatic and alicyclic amines, and heterocyclic bases, both mono- and bidentateCitation17,Citation18. Among them, pyrazole derivatives (substituted pyrazole ring as well as pyrazole ring containing bidentate N,N-donor ligands) were shown to be useful in complexing of transition metal ions, and the obtained complexes were proved to possess cytotoxic activityCitation19–21. The pyrazole ring containing moieties, especially with an additional group such as hydroxo or oxo, is of interest not only because of its chelating properties. Compounds such as 5-hydroxypyrazole and its derivatives may exist in several tautomeric forms, and this phenomenon has been extensively studiedCitation22,Citation23. The effect of the solvent and an additional heterocyclic part of the compound on the equilibrium between possible tautomers seems to be especially interesting.
This article reports the synthesis of new complexes of 1H-(2′-pyridyl)-3-methyl-5-hydroxypyrazole and 1H-(2′-pyridyl)-3-phenyl-5-hydroxypyrazole with palladium(II) ions. The obtained complexes were characterized by elemental analysis, and infrared (IR) and 1H nuclear magnetic resonance (NMR) spectroscopy. Crystals suitable for X-ray spectroscopy were also obtained, and their crystallographic structures are reported. The cytotoxic activity of the complexes against three cancer cell lines (HL-60 and NALM-6, human leukemia cell lines, and WM-115, a human melanoma cell line) was examined and compared to the activity of the ligands, as well as cisplatin and carboplatin as reference compounds. In addition, 1H NMR and density functional theory (DFT) studies of the ligands’ tautomerism are reported, and the influence of the heterocyclic ring on tautomeric equilibrium is discussed with reference to the results of other authors.
Experimental
Chemical materials and physical properties measurement
Reagent grade 2-hydrazinopyridine and bis(benzonitrile)dichloridopalladium(II) were purchased from Sigma Aldrich. Reagent grade methyl acetoacetate and ethyl benzoylacetate were purchased from Merck. All these compounds were used without further purification. All solvents (chloroform, methanol, ethanol, acetic acid, diethyl ether) were puriss. p.a. or equivalent purity grade and used without further purification.
Melting points were determined using Buchi Melting Point B-540 apparatus and reported without correction. The 1H and 13C NMR spectra were recorded on a Varian Mercury spectrometer (300 MHz). The IR spectra were registered in KBr on FT-IR Infinity MI-60 apparatus (Mattson), and elemental analyses of new compounds were obtained using a PerkinElmer PE 2400 CHNS analyzer in the Department of Bioorganic Chemistry (Medical University, Lodz).
Synthesis of compounds
Synthesis of 1H-(2′-pyridyl)-3-methyl-5- hydroxypyrazole (1): LCitation1
2-Hydrazinopyridine (218 mg, 2 mmol) was dissolved in absolute ethanol (10 cm3). Methyl acetoacetate (0.21 cm3, 2 mmol) was added to the solution and acetic acid (0.1 cm3) was added to the mixture. The mixture was heated under reflux for 9 h and left for 24 h at room temperature. The solvents were removed under reduced pressure using a rotational evaporator. The light-yellow oil residue was purified by “dry-column” flash chromatographyCitation24 using mixtures of chloroform and methanol (containing increasing amounts of methanol: 0%, 5%, 10%, etc.). Fractions containing the desired product, as checked by thin layer chromatography (TLC), were collected, and the solvents were evaporated under reduced pressure. The residue was dissolved in ethanol (3 cm3) under reflux, and after cooling to room temperature was left in the refrigerator. After 1 week the light-yellow solid crystals were filtered off, washed with diethyl ether, dried under reduced pressure, and used for further reactions and analyses (yield: 112 mg, 32.8%). M.p.: 108.5–109.7°C; νmax (KBr)/cm−1 3414, 1626, 1582, 1562, and 1469; δH (300 MHz; CDCl3; Me4Si) 2.26 (3H, s, CH3), 5.43 (1H, s, C4 pyrazole), 7.09–8.24 (4H, m, Py), 12.75 (1H, br s, OH/NH); δC (75.46 MHz; CDCl3; Me4Si) 15.0, 17.5, 43.4, 88.5, 111.9, 119.6, 139.9, 145.1, 151.7, and 157.0.
Synthesis of 1H-(2′-pyridyl)-3-phenyl-5- hydroxypyrazole (2): LCitation2
2-Hydrazinopyridine (370 mg, 3.4 mmol) was dissolved in absolute ethanol (10 cm3). Ethyl benzoylacetate (0.5 cm3, 2.88 mmol) was added, and finally acetic acid (1 cm3) was added to the mixture. The mixture was heated under reflux for 8 h and, after cooling to room temperature, left for 2 days in the refrigerator. Brown solid crystals were filtered off. Next they were dissolved in chloroform and purified by “dry-column” flash chromatography using mixtures of chloroform and methanol (containing increasing amounts of methanol: 0%, 5%, 10%, etc.). Fractions containing the desired product, as checked by TLC, were collected, and the solvents were evaporated under reduced pressure. The residue was recrystallized from ethanol, washed with diethyl ether, and dried under reduced pressure. Light-brown crystals were obtained (yield: 444 mg, 64.8%). M.p.: 117.4–118.9°C; νmax (KBr)/cm−1 3410, 1598, 1578, 1536, 1492, and 1467; δH (300 MHz; CDCl3; Me4Si) 5.96 (1H, s, C4 pyrazole), 7.14–8.30 (9H, m, Ph and Py), 12.84 (1H, br s, OH/NH); δC (75.46 MHz; CDCl3; Me4Si) 56.7, 61.3, 96.1, 98.7, 117.6, 124.4, 125.9, 133.6, 135.0, 137.4, 154.4, and 155.0.
Synthesis of PdLCitation1Cl2 (3)
1H-(2′-pyridyl)-3-methyl-5-hydroxypyrazole (1) (35.4 mg, 0.2 mmol) was dissolved in chloroform (2.5 cm3) and methanol (2.5 cm3). Then, a solution of Pd(C6H5CN)2Cl2 (76.7 mg, 0.2 mmol) in the same solvents was added dropwise with constant stirring. The mixture was left for 3 h on the magnetic stirrer, and left connected to the flask with diethyl ether for a week. As the diethyl ether slowly evaporated into the solution, brown-orange crystals began to precipitate. The solid was filtered off, washed with diethyl ether, and dried under reduced pressure (yield: 38.7 mg, 54.9%). Anal. for C9H9N3OPdCl2: calcd.: C, 30.7; H, 2.6; N, 11.9%; found: C, 30.7; H, 2.5; N, 11.6%. M.p.: does not melt to 400°C; νmax (KBr)/cm−1 3413, 1611, 1586, 1529, and 1481; δH (300 MHz; DMSO; Me4Si) 2.42 (3H, s, CH3), 5.47 (1H, s, C4 pyrazole), 7.38–8.83 (4H, m, Py).
Synthesis of PdLCitation2Cl2 (4)
To a solution of 1H-(2′-pyridyl)-3-phenyl-5-hydroxypyrazole (2) (51.4 mg, 0.22 mmol) in chloroform (2.5 cm3) and methanol (2.5 cm3), a solution of Pd(C6H5CN)2Cl2 (83.2 mg, 0.22 mmol) in the same solvents was slowly added with constant stirring. The mixture was left on the magnetic stirrer for an additional 3 h at room temperature. Next, the solvents were allowed to slowly evaporate for several days. The precipitated solid was filtered off, washed with diethyl ether, and dried under reduced pressure (yield: 71.9 mg, 78.8%). Anal. for C14H11N3OPdCl2·2/3CH3OH: calcd.: C, 40.0; H, 3.1; N, 9.55%; found: C, 40.0; H, 3.0; N, 9.5%; M.p.: 295–296°C; νmax (KBr)/cm−1 3410, 1611, 1583, 1567, 1520, 1480; δH (300 MHz; DMSO; Me4Si) 5.63 (1H, s, C4 pyrazole), 7.02–8.80 (9H, m, Ph and Py).
Obtaining crystals of complexes 3 and 4 suitable for X-ray analysis
In the case of complex 3, crystals suitable for X-ray analysis were obtained directly from the synthesis of this complex. In the case of complex 4, a small amount (5 mg) was dissolved in a 1:1 mixture of chloroform and methanol (5 cm3) and the diethyl ether was allowed to slowly evaporate into the solution. After 2 weeks the resulting crystals were filtered off, washed with diethyl ether, and dried under reduced pressure.
Quantum calculations
All calculations were performed using the Gaussian 03 program package, revision D.01Citation25.
Calculation of compounds 1 and 2 tautomers’ structures and energies
First, scans of the potential energy surface with rotation around the bond between pyrazole and pyridine rings of compounds 1 and 2 were performed using the semiempirical AM1 methodCitation26. In the case of compound 2, an additional scan of energy changes with rotation of the bond between pyrazole and phenyl rings was performed. The procedure was repeated for all three tautomers of each compound. The resulting structures corresponding to the energy minima were optimized at the DFT level, using the Becke three-parameter exchange functional with the Lee–Yang–Parr correlation functional (B3LYP)Citation27 and 6-31+G(d,p) basis setCitation28–30. Optimization was carried out in the gas phase, chloroform, dimethylsulfoxide (DMSO), and methanol. In the case of optimization in solvent, the polarized continuous model of the solvent based on the integral equation formalism (IEFPCM)Citation31–35 was used. Vibrational analyses were performed to identify the obtained structures as stationary points and to determine the zero-point energy corrections and thermal corrections to Gibbs free energies. In the case of the two enantiomeric rotamers, only one structure was optimized, but two different rotamers with identical energies were taken into account in further calculations. The obtained structures were visualized using the MercuryCitation36,Citation37 program provided by the Cambridge Crystallographic Data Centre (UK).
Calculation of compounds 3 and 4 structures
The structures of the complexes were optimized at the DFT level using the Becke three-parameter exchange functional with Lee–Yang–Parr correlation functional (B3LYP). For the Pd atom, the SDD (Stuttgart group) effective core potential for core electrons with SDD basis for valence electrons was usedCitation38. For the other atoms, the 6-31+G(d,p) basis set was used. Vibrational analyses were performed to identify the obtained structures as stationary points. Visualizations of structures were provided using the Mercury program.
Calculation of tautomers’ population distribution
The amount of each tautomer in the gas phase and in solution was calculated using the Boltzmann distribution law. The energies used in the calculations were the enthalpy and Gibbs free energy obtained from quantum calculations. To take into account the distribution of each tautomer over multiple energy states (rotamers), the ratio of each tautomer to another was calculated from:
where: N is the number of tautomer molecules, q is the partition function for the tautomer, ϵ is the energy corresponding to the tautomer’s lowest energy state, kb is the Boltzmann constant, and T is the temperature.
The partition function of each tautomer was calculated from:
where: ϵi is the energy of rotamer i and ϵ0 is the energy corresponding to the tautomer’s lowest energy stateCitation39.
The percentage of each tautomer was obtained with the assumption that all three tautomers give 100% of compound forms in the system.
X-ray structure determination of 3 and 4
X-ray data were collected at 200 K with MoKa radiation (l = 0.71073 Å) using an Oxford XCalibur diffractometer (3) and a Nonius KappaCCD diffractometer (4). The structures were solved using direct methodsCitation40 and refined with SHELXL-97 by full-matrix least-squares on FCitation2 Citation41. Two disordered methanol molecules in 4 were handled by split models and refined isotropically. All other non-hydrogen atoms were refined anisotropically. The crystal data and X-ray details are given in Table S1 (“Supplementary material”). Further details are available from the Crystallographic Data Centre under the depository numbers CCDC 695754 (3) and 695755 (4). Copies of the data can be obtained free of charge upon application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (e-mail: [email protected]).
Cytotoxicity assay
Cell cultures
Human skin melanoma WM-115 cells as well as human leukemia promyelocytic HL-60 and lymphoblastic NALM-6 cell lines were used. Leukemia cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and antibiotics (100 μg/ml streptomycin and 100 U/ml penicillin). For melanoma WM-115 cells, Dulbecco’s minimal essential medium (DMEM) instead of RPMI 1640 was used. Cells were grown at 37°C in a humidified atmosphere of 5% CO2 in air.
Cytotoxicity assay by MTT
The cytotoxicity of ligands 1 and 2, complexes 3 and 4, cisplatin, and carboplatin was determined by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Sigma, St. Louis, MO) assay as described elsewhereCitation19. Briefly, after 46 h of incubation with drugs, the cells were treated with the MTT reagent and incubation was continued for 2 h. MTT–formazan crystals were dissolved in 20% sodium dodecyl sulfate (SDS) and 50% dimethyl formamide (DMF) at pH 4.7, and absorbance was read at 562 and 630 nm on an enzyme-linked immunosorbent assay (ELISA) plate reader (ELX 800; Bio-Tek, VT). The values of IC50 (concentration of test compound required to reduce the cells’ survival fraction to 50% of control) were calculated from concentration–response curves and used as a measure of cellar sensitivity to a given treatment. Complexes and cisplatin were tested for their cytotoxicity in a final concentration 10−7–10−3 M. As a control, cultured cells were grown in the absence of drugs. Data points represent means of at least 12 repeats ± SD.
Results and discussion
Synthesis of compounds 1 and 2: NMR and DFT study of 1 and 2 tautomeric forms
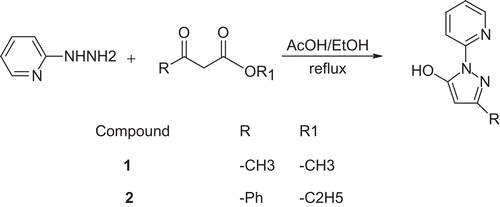
Synthesis of compounds 1 and 2 was done as shown in , based on methods reported previouslyCitation42–45 (for details see “Experimental” section). Products were purified using the “dry-column” flash chromatography method. As a product, light-yellow (compound 1) or light-brown (compound 2) crystals were obtained.
Scheme 1. Synthesis of compounds 1 and 2.
There are three possible tautomeric forms of both compounds 1 and 2Citation46 as shown in
Scheme 2. Tautomeric forms of compounds 1 and 2.
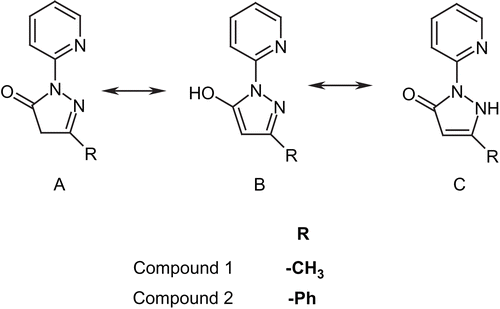
The tautomeric forms of pyrazole derivatives have been extensively studied in the pastCitation47,Citation48. It has been shown that, in general, form A in non-polar solvent is the most stable. With an increase of solvent polarity, the amount of form C increases and predominates in aqueous solution, with a lower amount of form BCitation49. The pyridine ring in compounds 1 and 2 includes a nitrogen atom with lone electron pair. This is important, because it can be involved in an intramolecular hydrogen bond creation that would stabilize the B and C tautomers even in non-polar solvents.
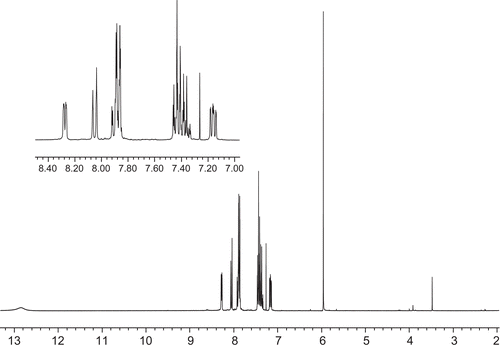
In order to characterize occurrences of the compound 1 and 2 tautomers in solution, 1H NMR analyses were conducted. The spectra acquired in chloroform are depicted in and .
Figure 1. 1H NMR spectrum of compound 1.
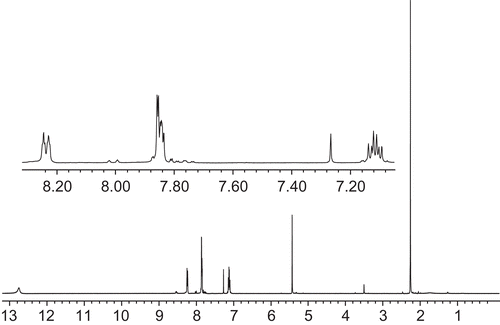
Figure 2. 1H NMR spectrum of compound 2.
Almost all signals visible in the 1H NMR spectrum of compound 1 may be attributed to the corresponding protons. The strong singlet signal at 2.26 ppm comes from the methyl group protons (3H). The singlet at 5.43 may be attributed to the methine group proton (1H) of tautomer B or C. In the “aromatic” region of the spectrum, three sets of signals exhibiting complex patterns are observable. First at 7.09–7.15 is the signal from the proton attached to the C5 pyridine ring (1H). The next signal at 7.83–7.86 comes from two protons attached to C3 and C4 of the pyridine ring (2H). At 8.23–8.24 is a signal of the C6 pyridine ring proton (1H). The broad signal at 12.75 ppm comes from the proton attached to the oxygen in the hydroxyl group of tautomer B or the nitrogen in tautomer C. These data and signal assignment are in accordance with data reported previouslyCitation44,Citation46. Of interest is the small signal at 3.50 ppm. It may be assigned to protons of the methylene group from an alternative tautomeric form (A) of compound 1. The intensity of this signal compared to the signal of the methine group proton is in the ratio 1:7, which suggests that in chloroform the tautomer B or C of compound 1 is favorable.
The 1H NMR spectrum of compound 2 reveals some similarity to the spectrum presented previously. The main differences between this and previous spectra are as follows. There is a lack of methyl group protons’ signal: instead the additional signals from phenyl group protons are observable in the “aromatic” region (7.2–8.1 ppm); the signal of the methine group proton (tautomers B and C) is shifted to 5.96 ppm; the signal of the methylene group protons (tautomer A) is shifted to 3.91 ppm; the signal of the hydroxyl group or NH proton is shifted to 12.84 ppm; and signals from pyridine protons are slightly shifted to more deshielded values. Also, the ratio of the signal of the methylene group protons of tautomer A to the signal of the methine group proton is changed to 1:40.
Additionally, 1H NMR and 13C NMR spectra of compounds 1 and 2 in DMSO and compound 1 in methanol were obtained in order to check the influence of solvent polarity on the equilibrium between tautomeric forms of these compounds. Due to the poor solubility of compound 2 in methanol, the 1H NMR and 13C NMR spectra were not obtained. The percentages of tautomer A and tautomers B/C in solution, calculated based on ratios of the signal strength of the methine group proton of tautomers B/C to the signal strength of the tautomer A methylene group protons in 1H NMR spectra of both compounds in different solvents, are summarized in .
Table 1. Percentage of tautomeric forms in solution calculated from 1H NMR.
Information obtained from 13C NMR spectra does not allow quantitative analysis of the tautomeric form present in each solution. However, it can be used as confirmation of the presence of A and B/C tautomeric forms of compounds 1 and 2 in chloroform.
In the 13C NMR spectrum of 1 in chloroform, signals of the methyl group carbon of tautomers B/C (15.0 ppm) and tautomer A (17.5 ppm) are visible. The C4 carbon of the pyrazole ring gives a signal at 43.4 ppm for tautomer A and 88.5 ppm for tautomers B/C. The lack of two signals in the 10–20 ppm region of the spectra and lack of a signal around 40 ppm in DMSO or methanolic solution indicates that the amount of form A of 1 is very small or that it does not exist in these conditions. Analysis of the 13C NMR spectra of compound 2 gives similar results. In chloroform, two signals from the C4 carbon are visible (56.7 ppm for tautomer A and 98.7 ppm for tautomers B/C). The lack of this tautomer A signal in DMSO and occurrence of the signal of tautomers B/C (shifted to 85.5 ppm) indicates that the main forms in DMSO are the tautomers B and C of compound 2.
As the signals of the methine group protons of tautomeric forms B and C are indistinguishable in 1H NMR and quantitative analysis is impossible, quantum calculations were carried out in order to reveal the percentage of each tautomer in equilibrium in different solvents. First, scan of the rotation around the bond linking the rings was done using the semiempirical AM1 method as implemented in the Gaussian 03 program for all tautomers. Optimizations of the found conformers’ structures were provided in the gas phase at the DFT level using the B3LYP functional and 6-31+G(d,p) basis set as implemented in the Gaussian 03 program. The minimum energy was confirmed by vibrations calculation (lack of imaginary vibrations confirms the energetic minimum). These calculations were repeated using the polarizable continuous model of the solvent (PCM). The simulated solvents were chloroform, DMSO, and methanol, as used during obtaining the 1H NMR spectra (for details and references concerning calculations see “Experimental” section). The structures of the lowest energy conformer for each tautomer in the gas phase are shown in and . Dihedral angles between adjacent rings are revealed.
Figure 3. Calculated structures of the lowest energy conformers of each tautomer of 1 in the gas phase with selected dihedral angles (°).
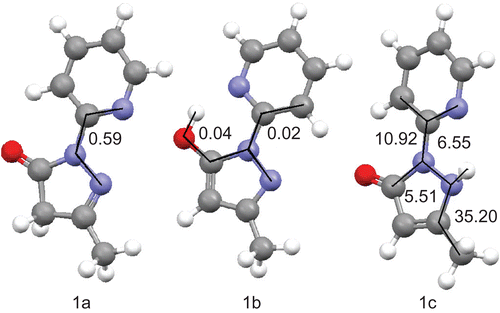
Figure 4. Calculated structures of the lowest energy conformers of each tautomer of 2 in the gas phase with selected dihedral angles (°).
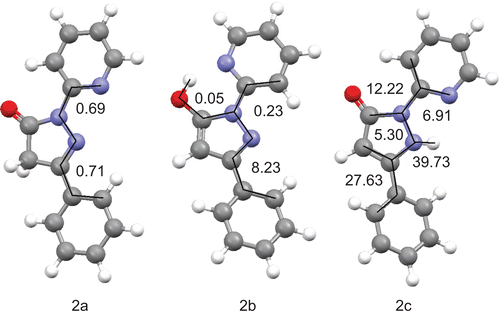
After the enthalpy and free Gibbs energy of each conformer were calculated, the theoretical amount of each tautomer was estimated based on the Boltzmann distribution law. Results are shown in and . Details of calculations of each tautomer percentage are described in the “Experimental” section.
Table 2. Enthalpy and percentage of tautomers calculated at B3LYP/6-31+G(d,p) level of theory.
Table 3. Gibbs free energy and percentage of tautomers calculated at B3LYP/6-31+G(d,p) level of theory.
The most stable tautomer of 1 and 2 as calculated without the influence of solvent (in the gas phase) is the B (5-hydroxo) form. However, in solution with an increase of polarity of solvent the amount of tautomer C increases. The calculated percentage of the A tautomeric form remains at almost the same level, except for compound 2 in DMSO solution, in which case the amount of 2A rapidly increases when the Gibbs free energy is taken into account. Calculations based upon the Gibbs free energy tend to underestimate the amount of 1A in all solvents and overestimate the amount of 2A in DMSO and chloroform. Calculations based on the enthalpy underestimate the percentage of tautomer A for both compounds, with the exception of the amount of tautomer 2A in DMSO. However, both calculations predict correctly that tautomers B and C are more stable than tautomer A. Additionally, prediction of the most stable tautomer of 2 in the gas phase is in agreement with the crystal structure of this compound reported by other authorsCitation45. However, this result should be viewed with caution, as single-molecule gas-phase calculations do not take into account the intermolecular hydrogen bonding that may stabilize tautomer 2B in the solid state. Based on all the results, it can be stated that in solution, together with an increase of polarity of the solvent, the amount of tautomer C increases and becomes the main form present in solution in solvents such as DMSO and methanol.
A comparison with previous studies concerning similar pyrazole derivatives substituted on N1 with the phenyl ring reveals the importance of heterocyclic nitrogen. It possesses a free pair of electrons and may be included in hydrogen bonding. Such an interaction is able to lower the compound’s energy and stabilize tautomers capable of generating intramolecular H-bonding (tautomers B and C). In fact, the results of previous authors reveal that in the case of 3-methyl-1-phenylpyrazolin-5-one and 1,3-diphenylpyrazolin-5-one the main form in chloroform solution is the A tautomer. In the case of the former compound, in a more polar solvent such as DMSO the amount of tautomer A decreases to about 20%, and in D2O the main occurring form is tautomer CCitation49,Citation50. Results presented in this article reveal that even in chloroform, tautomers B and C are the main forms of compounds 1 and 2. The reason for this difference is the ability of 1 and 2 to generate intramolecular H-bonding. As this is only possible in tautomeric forms B and C, hence these two forms are energetically privileged over form A. Methods used in this study do not allow distinction between tautomers B and C. However, the calculations show that with an increase of polarity of the solvent the amount of tautomer C also increases. This is in agreement with the statement of authors of other publication that in the case of 1H-(2′-pyridyl)-3-methyl-5-hydroxypyrazole, the main form in solution is tautomer CCitation46.
Synthesis of complexes
The complexes of compounds 1 and 2 with palladium(II) ions were obtained in reactions with bis(benzonitrile)dichloridopalladium(II) in chloroform/methanol solution as shown in
Scheme 3. Synthesis of complexes 3 and 4.
Complexes were obtained as brown-orange solids. Solids were filtered off, washed with diethyl ether, and dried under reduced pressure. Purity of the obtained complexes was checked by TLC chromatography and elemental analysis. Next, the obtained complexes were used for further studies.
X-ray structure analysis and DFT calculations of Pd(II) complexes 3 and 4
Single crystals of 3 suitable for X-ray structure analysis were obtained by isothermic diffusion of diethyl ether vapor into the chloroform/methanol solution of 3. Compound 3 crystallizes with one molecule of CH3OH originating from the synthesis of 3. The crystallographic data and details of structure refinement for complex 3 are summarized and given in the “Supplementary material” (Table S1). The molecular structure of 3 is shown in , and selected bond lengths and angles are given in the “Supplementary material” together with the values from DFT calculations (Table S2).
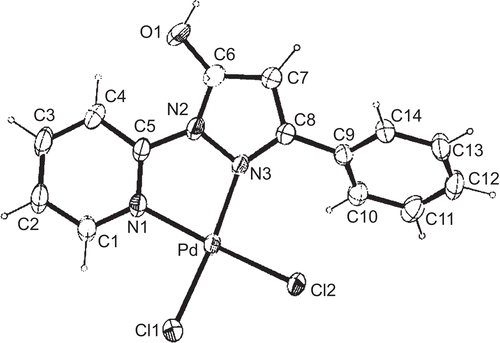
Figure 5. ORTEP view of molecular structure of Pd(II) compound 3 with atom numbering scheme and ellipsoids at 30% probability level.
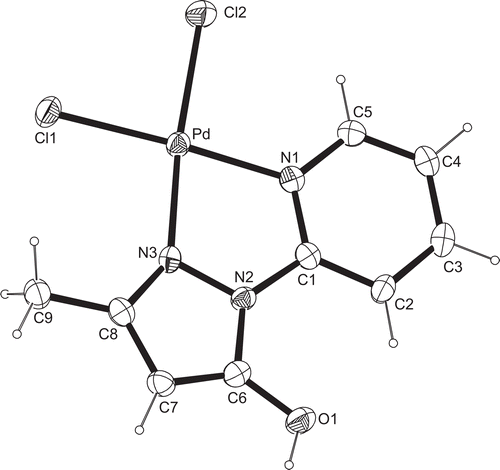
In complex 3, 1H-(2′-pyridyl)-3-methyl-5-hydroxypyrazole coordinates as a bidentate ligand with palladium(II), resulting in a cis-position of both chlorido ligands. The configuration at the Pd-center is distorted square-planar, and the sum of angles around Pd is exactly 360°. Both the Pd–Cl bonds (Pd–Cl1 2.2799(9), Pd–Cl2 2.3000(9) Å) and the shorter Pd–N bonds (Pd–N1 2.029(2), Pd–N3 2.017(2) Å) differ only slightly. The angle N1–Pd–N3 (80.57(9)°) of the chelate ligand is the smallest, then follows the angle Cl1–Pd–Cl2 (88.33(3)°), whereas the two angles Cl1–Pd–N1 (94.25(6)°) and Cl2–Pd–N3 (96.94(7)°) are significantly larger than 90°. This may be due to the small angle of the N,N9-chelate system and corresponds to values found earlier in a similar Pd-complexCitation51 and also in complex 4. The trans ligands form angles Cl1–Pd–N1 and Cl2–Pd–N3 of about 175°. The five- and six-membered rings of pyridyl–pyrazole lie in the same plane together with the substituents (O9 and C9) at the pyrazole. The maximum deviation from planarity is encountered between the N2–N3–C8–C7–C6 five-membered ring and the N1–C1–C2–C3–C4–C5 six-membered ring, and amounts to only 5.71(16)°. In the structure, the intermolecular hydrogen bonds can be observed. The first, between the OH group and O2 oxygen of the methanol molecule possess a length of 2.543(3) Å. The O1–H1 O2 angle amounts to 156.4°. The other two hydrogen bonds are O2–H2 Cl2 (length 3.323 Å; angle 156°) and O2–H2 Cl1 (length 3.282 Å; angle 127°).
Single crystals of 4 suitable for X-ray structure analysis were obtained by isothermic diffusion of diethyl ether vapor into the solution of 4 in chloroform/methanol. Compound 4 crystallizes with one disordered CH3OH-molecule originating from the synthesis of 4. The crystallographic data and details of structure refinement for complex 4 are summarized and given in the “Supplementary material” (Table S1). The molecular structure of 4 is shown in , and selected bond lengths and angles are given in the “Supplementary material” together with the values from DFT calculations (Table S3).
Figure 6. ORTEP view of molecular structure of Pd(II) compound 4 with atom numbering scheme and ellipsoids at 30% probability level.
The monomeric complex 4 contains the bidentate pyridyl substituted pyrazole ligand 2 and two chlorido ligands in the cis-position. Compound 4 exhibits a slightly distorted square-planar configuration at the Pd(II)-center; the sum of angles around Pd is 360°. Both the Pd–Cl bond (Pd–Cl1 2.2828(13), Pd–Cl2 2.2822(12) Å) and the Pd–N bond lengths (Pd–N1 2.033(3), Pd–N3 2.037(3) Å) are equal. While the angle N1–Pd–N3 (80.62(13)°) of the chelate system is the smallest and the angle Cl1–Pd–Cl2 (88.5(4)°) is almost 90°, the angles Cl1–Pd–N1 (93.71(10)°) and Cl2–Pd–N3 (97.51(9)°), however, are not equal and are significantly larger than 90°. This is due to the small angle of the N,N9-chelate ligand.
The five- and six-membered rings of pyridyl–pyrazole ligand 2 lie almost in the same plane, together with the atoms O1 and C9 of the substituents (C6 and C8) at the pyrazole, as well as with the PdCl2 moiety. The maximum deviation amounts to 9.5(3)°. The higher deviation value with comparison to compound 3 is probably due to the steric hindrance of the phenyl substituent. The phenyl substituent at atom C8 is bent out from this molecular plane by only 43.8(3)°. The relevant dihedral angle is smaller than in the similar Pt-complex with a phenolic substituent (~70°)Citation52, because of less steric hindrance. In the structure, an intermolecular hydrogen bond between compound 4 and the disordered methanol molecule can be observed. The length of this bond is 2.666 Å and the angle (O1–H1 O2) value is 166.7°. These values are in agreement with those found for compound 3.
DFT calculations of complexes 3 and 4 structures were also carried out (for details see “Experimental” section). Comparisons of selected bond lengths as well as angle values obtained in X-ray analyses and DFT calculations are shown in Tables S1 and S2 (“Supplementary material”). Comparisons of complex structures obtained by theoretical calculations and X-ray analysis are presented in and . As can be seen from these figures and the error values presented in the tables mentioned above, the calculated structures are in good agreement with structures obtained experimentally. In the case of both complexes, the main problem that occurs is overestimation of Pd–N bond lengths, which are calculated to be too long by 0.05–0.07 Å. Also, the computed angles around the palladium ion are slightly different from those obtained in X-ray analysis. The N–Pd–N angles tend to be underestimated and Cl–Pd–Cl angles are overestimated. However, the overall distorted square-planar configuration around the metal ion is reproduced quite well. Other differences are overestimation of the C–O bond (0.02 Å) and overestimation of C–C bonds in pyridine and benzene rings (0.02 Å). The phenyl substituent in compound 4 is bent out in the other direction in comparison to the X-ray structure. However, because of the planarity of other parts of the compound, this is not an issue. The dihedral angle that characterizes this bending differs only by 3°, which is not a large difference if the weak nature of interactions is taken into account.
Figure 7. Comparison of compound 3 structures from X-ray crystallography (gray scale) and DFT calculations (black).
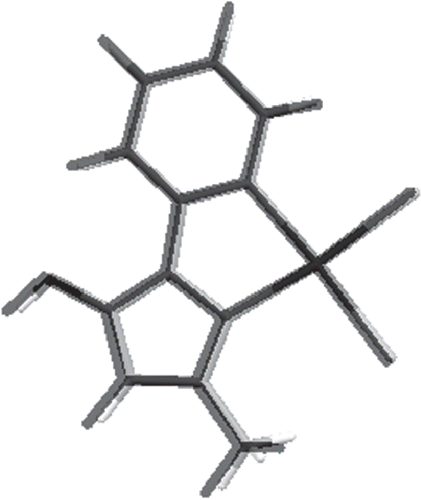
Figure 8. Comparison of compound 4 structures from X-ray crystallography (gray scale) and DFT calculations (black).
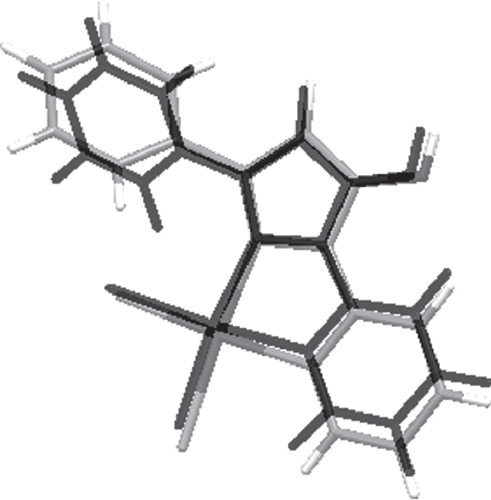
Cytotoxic activity
The cytotoxic activity of ligands 1 and 2 and complexes 3 and 4 was examined on three cancer cell lines (HL-60 and NALM-6 human leukemia cell lines, and WM-115 human melanoma cell line), and with the activity of cisplatin and carboplatin as reference compounds. The IC50 values of tested compounds are collected in . The choice of cancer cell lines was influenced by the fact that both leukemia and melanoma are still serious therapeutic problems and new active agents are needed.
Table 4. IC50 values of tested compounds.
As can be seen from the collected data, the ligands themselves exhibit high cytotoxic activity. Both ligands are more active against leukemia cell lines. Compound 2 possesses higher (than 1) activity against leukemia cell lines, while compound 1 is more active (than compound 2) against the melanoma cell line. The complex of 1 with the palladium(II) ion (compound 3) shows lower activity than the ligand against the leukemia cell lines and comparable activity against the melanoma cell line. This is in contrast with compound 4 (complex of 2 with palladium(II) ion), which reveals comparable activity to the ligand against the leukemia cell lines and three times higher activity against the melanoma cell line. In the case of the HL-60 leukemia cell line, compounds 2 and 4 possess values of IC50 similar to that of carboplatin. In the case of the second leukemia cell line (NALM-6), all compounds are less active than the reference compounds. For the WM-115 melanoma cell line, tested compounds are less cytotoxic than cisplatin, but more active than carboplatin. The highest activity in this case is exhibited by 4, which is almost eight times more active than carboplatin and only three times less active than cisplatin. Modifications of this compound may lead to a new therapeutic agent for the treatment of melanoma.
Conclusions
In this article, synthesis, characterization, structure determination, and cytotoxic activity studies of Pd(II) complexes with substituted pyridyl–pyrazole ligands have been reported. Additionally, studies of the ligands’ tautomers existence in different solvents have also been provided.
Based upon the results of crystallographic studies, it can be stated that both ligands act as bidentate chelators. The Pd complexes acquire square-planar geometry, which is in agreement with expectations. In the case of the 3-methyl-substituted pyrazole derivative, synthesis of both the ligand and the complex was difficult. This can be seen from the reaction yields, which are poor in the former case and only fair in the latter. Synthesis of the 3-phenyl pyrazole derivative and its complex was easier and gave better yields.
1H NMR studies of the existence of tautomeric forms in different solvents have shown that the main forms of both ligands in solution are tautomers B and C. Additionally quantum calculations revealed that with increasing polarity of the solvent the amount of tautomeric form C of both ligands also increases, and that this form is preferred over form B. However, the energy of tautomer B is the lowest when calculated in the gas phase, and this is in accordance with the form of ligand 2 in the solid state.
Comparison of the crystal structures of complexes 3 and 4 with structures calculated on the DFT level show a good agreement of data. The main problem is the slight distortion of geometry around the Pd atom. This may be caused by the use of an effective core potential to describe the Pd atom in the calculations (see “Experimental” section). However, this distortion does not cause major changes in molecule geometry. Based on these results, it can be stated that the method used in the calculations is suitable for obtaining the general structure of similar types of compounds.
The results for cytotoxic activity are somewhat ambiguous. The ligands reveal high toxicity by themselves. Complexing with the palladium ion does not increase the activity of ligand 1. Results of studies on the 3-phenyl-substituted pyrazole derivative are much more promising. In the case of the HL-60 leukemia cell line, compound 4 is almost as active as carboplatin. Unfortunately, the activity of the ligand is the same. However, in the case of the melanoma cell line the results are much better. The activity of ligand 2 is moderate, but the activity of complex 4 is three times greater. This gives hope that further modifications of the structure of complex 4 may lead to an efficient agent against melanoma.
Acknowledgements
Financial support from the Ministry of Science and Higher Education (Research Grant No. N N405 428434 to EB), the award of a DAAD fellowship (No. A/07/73915 to MC), and access to supercomputing facilities at AGH Cyfronet Krakow (Grant No. MNiSW/SGI3700/UMLodz/004/2008 to MC) are gratefully acknowledged.
Declaration of interest: The authors report no conflicts of interest.
References
- Rosenberg B, VanCamp L, Trosko JE, Mansour VH. Nature 1969;222:385–6.
- Rosenberg B, VanCamp L. Cancer Res 1970;30:1799–802.
- Desoize B, Madoulet C. Crit Rev Oncol Hematol 2002;42:317–25.
- Lebwohl D, Canetta R. Eur J Cancer 1998;4:1522–34.
- Clarke MJ, Zhu F, Frasca DR. Chem Rev 1999;99:2511–33.
- Van den Berg JH, Beijnen JH, Balm AJM, Schellens JHM. Cancer Treat Rev 2006;32:390–7.
- Carotti S, Guerri A, Mazzei T, Messori L, Mini E, Orioli P. Inorg Chim Acta 1998;281:90–4.
- Clarke MJ. Coordin Chem Rev 2002;232:69–93.
- Alessio E, Mestroni G, Bergamo A, Sava G. Curr Top Med Chem 2004;4:1525–35.
- Daniel KG, Gupta P, Harbach RH, Guida WC, Dou QP. Biochem Pharmacol 2004;67:1139–51.
- Cini R, Tamasi G, Defazio S, Hursthouse MB. J Inorg Biochem 2007;101:1140–52.
- De Vizcaya-Ruizi A, Riveiro-Muller A, Ruiz-Ramirez L, Kass GEN, Kelland LR, Orr RM, et al. Toxicol In Vitro 2000;14:1–5.
- Butour JL, Wimmer S, Wimmer F, Castan P. Chem-Biol Interact 1997; 104:165–78.
- Budzisz E, Keppler BK, Giester G, Wozniczka M, Kufelnicki A, Nawrot B. Eur J Inorg Chem 2004;22:4412–19.
- Gao E-J, Wang K-H, Gu X-F, Yu Y, Sun Y-G, Zhang W-Z, et al. J Inorg Biochem 2007;101:1404–9.
- Zhao G, Sun H, Lin H, Zhu S, Su X, Chen Y. J Inorg Biochem 1998;72:173–7.
- Wong E, Giandomenico ChM. Chem Rev 1999;99:2451–66.
- Zhang CX, Lippard SJ. Curr Opin Chem Biol 2003;7:481–9.
- Budzisz E, Krajewska U, Rozalski M, Szulawska A, Czyz M, Nawrot B. Eur J Pharmacol 2004;502:59–65.
- Onoa GB, Moreno V, Font-Bardia M, Solans X, Perez JM, Alonso C. J Inorg Biochem 1999;75:205–12.
- Chen R, Liu C-S, Zhang H, Guo Y, Bu X-H, Yang M. J Inorg Biochem 2007;10:412–21.
- Elguero J, Jacquier R, Tarrago G. B Soc Chim Fr 1967;10:3772–9.
- Katritzky AR, Maine FW, Golding S. Tetrahedron 1965;21:1693–9.
- Harwood LM. Aldrichim Acta 1985;18:25.
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian 03, revision D.01. Wallingford, CT: Gaussian, Inc., 2004.
- Dewar MJS, Zoebisch EG, Healy EF, Stewart JJP. J Am Chem Soc 1985;107:3902–9.
- Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. J Phys Chem 1994;98:11623–7.
- Hehre WJ, Ditchfield R, Pople JA. J Chem Phys 1972;56:2257–61.
- Hariharan PC, Pople JA. Theor Chim Acta 1973;28:213–22.
- Frisch MJ, Pople JA, Binkley JS. J Chem Phys 1984;80:3265–9.
- Miertus S, Scrocco E, Tomasi J. Chem Phys 1981;55:117–29.
- Miertus S, Tomasi J. Chem Phys 1982;65:239–45.
- Mennucci B, Tomasi J. J Chem Phys 1997;106:5151–8.
- Cancès E, Mennucci B, Tomasi J. J Chem Phys 1997;107:3032–41.
- Cossi M, Scalmani G, Rega N, Barone V. J Chem Phys 2002;117:43–54.
- Bruno IJ, Cole JC, Edgington PR, Kessler M, Macrae CF, McCabe P, et al. Acta Crystallogr B 2002;B58:389–97.
- Macrae CF, Edgington PR, McCabe P, Pidcock E, Shields GP, Taylor R, et al. J Appl Crystallogr 2006;39:453–7.
- Andrae D, Haussermann U, Dolg M, Stoll H, Preuss H. Theor Chim Acta 1990;77:123–41.
- Atkins P, De Paula J. Atkin’s Physical Chemistry, 8th edn. Oxford: Oxford University Press, 2006:560–87.
- Altomare A, Burla MC, Camalli M, Cascarano GL, Giacovazzo C, Guagliardi A, et al. J Appl Crystallogr 1999;32:115–19.
- Sheldrick GM. SHELXL-97, version 97-2. University of Gottingen, 1997.
- Watanabe K, Morinaka Y, Iseki K, Watanabe T, Yuki S, Nishi H. Redox Rep 2003;8:151–5.
- Sieler J, Kempe R. Z Kristallogr 1992;198:313–14.
- Nakagawa H, Ohyama R, Kimata A, Suzuki T, Miyata N. Bioorg Med Chem Lett 2006;16:5939–42.
- McFerrin CA, Hammer RP, Fronczek FR, Watkins SF. Acta Crystallogr E 2006;E62:o2518–19.
- Iyengar DS, Prasad KK, Venkataratnam RV. Tetrahedron Lett 1972;37: 3937–40.
- Elguero J, Jacqiuer R, Tarrago G. B Soc Chim Fr 1967;10:3772–9.
- Elguero J, Jacqiuer R, Tarrago G. B Soc Chim Fr 1967;10:3780–94.
- Katritzky AR, Maine FW. Tetrahedron 1964;20:299–314.
- Hawkes GE, Randall EW, Elguero J, Marzin CJ. J Chem Soc Perkin Trans 2 1977:1024–7.
- Budzisz E, Lorenz I-P, Mayer P, Paneth P, Szatkowski L, Krajewska U, et al. New J Chem 2008;32:2238–44.
- Budzisz E, Miernicka M, Lorenz I-P, Mayer P, Krajewska U, Rozalski M. Polyhedron 2009;28:637–45.