Abstract
In mammals, aging is linked to a decline in the activity of citrate synthase (CS; E.C. 2.3.3.1), the first enzyme of the citric acid cycle. We used 2,2′-azobis(2-amidinopropane) dihydrochloride (AAPH), a water-soluble generator of peroxyl and alkoxyl radicals, to investigate the susceptibility of CS to oxidative damage. Treatment of isolated mitochondria with AAPH for 8–24 h led to CS inactivation; however, the activity of aconitase, a mitochondrial enzyme routinely used as an oxidative stress marker, was unaffected. In addition to enzyme inactivation, AAPH treatment of purified CS resulted in dityrosine formation, increased protein surface hydrophobicity, and loss of tryptophan fluorescence. Propyl gallate, 1,8-naphthalenediol, 2,3-naphthalenediol, ascorbic acid, glutathione, and oxaloacetate protected CS from AAPH-mediated inactivation, with IC50 values of 9, 14, 34, 37, 150, and 160 μM, respectively. Surprisingly, the antioxidant epigallocatechin gallate offered no protection against AAPH, but instead caused CS inactivation. Our results suggest that the current practice of using the enzymatic activity of CS as an index of mitochondrial abundance and the use of aconitase activity as an oxidative stress marker may be inappropriate, especially in oxidative stress-related studies, during which alkyl peroxyl and alkoxyl radicals can be generated.
| Abbreviations: | ||
| AAPH, | = | 2,2′-azobis(2-amidinopropane) dihydrochloride |
| ANSA, | = | 8-anilino-1-naphthalenesulfonic acid |
| ASC, | = | ascorbate |
| BDE, | = | bond dissociation enthalpy |
| CS, | = | citrate synthase; diTyr, dityrosine |
| EGCG, | = | epigallocatechin gallate |
| ETC, | = | electron transport chain |
| GSH, | = | reduced glutathione |
| H2O2, | = | hydrogen peroxide |
| ND, | = | naphthalenediol |
| OAA, | = | oxaloacetate |
| PC-12AC, | = | adherent clone of the rat adrenal pheochromocytoma cell |
| PG, | = | propyl gallate |
| ROO•, | = | alkyl peroxyl radical |
| RO•, | = | alkoxyl radical |
| ROS, | = | reactive oxygen species |
Introduction
A large body of evidence exists showing that mitochondria are the major source of free radicals that contribute to cellular aging (for a recent review, see reference 1). Mitochondrial sources of free radicals include electron leakage or incomplete reduction of molecular oxygen by the mitochondrial electron transport chain (ETC), leading to the generation of reactive oxygen species (ROS) such as superoxide (O2•–), hydrogen peroxide (H2O2), alkyl peroxyl (ROO•), lipid peroxyl (LOO•), and hydroxyl (HO•) radicalsCitation2. As much as 1–2% of O2 used in mitochondrial respiration is converted to ROS, primarily by complexes I and III of the ETCCitation2. In addition, mitochondrial ROS are produced enzymatically by pyruvate, α-ketoglutarate, acyl-coenzyme A (CoA), isocitrate, and dihydrolipoyl dehydrogenases, as well as monoamine oxidase BCitation3–7. Located in close proximity to the sites of ROS production, the enzymes of the citric acid cycle are especially prone to oxidative attack. The most notable targets are aconitaseCitation8,Citation9 and α-ketoglutarate dehydrogenaseCitation5,Citation6,Citation10.
Citrate synthase (CS), the first enzyme of the citric acid cycle, catalyzes the irreversible condensation of oxaloacetate and acetyl-CoA to form citrate and CoA-SH. The enzyme is controlled by the availability of oxaloacetate (which is present in concentrations much less than 1 μM in the mitochondria) as well as by allosteric inhibition by NADH (reduced nicotinamide adenine dinucleotide), ATP (adenosine triphosphate), and succinyl-CoA (all products of the citric acid cycle). The binding of oxaloacetate induces a conformational change in the enzyme that facilitates the binding of acetyl-CoA. CS is found in both prokaryotes and eukaryotes, and in the latter, it is located exclusively in the mitochondrial matrixCitation11.
CS activity has been widely used as an indicator of cellular mitochondrial content, function, and integrityCitation12–15 (for a review, see reference 16). However, recent evidenceCitation17 suggests that changes in CS activity do not parallel changes in mitochondrial DNA content and other mitochondrial respiratory enzymes. Similarly, the increase in CS activity during postnatal development did not correlate with that of cytochrome c oxidase, a key enzyme of mitochondrial respirationCitation18. That, along with the possible susceptibility of CS to oxidative modifications investigated here, may limit the use of CS enzyme activity as an indicator of mitochondrial abundance or integrity.
CS activity is significantly reduced in the brains and livers of old mice, with concomitant age-dependent increases in oxidative stress parameters (such as products of lipid peroxidation and activities of antioxidant enzymes (e.g. superoxide dismutases))Citation19. Aged rats also display a decline in CS activityCitation20. Decreased CS activity was noticeable in rat brain tumors where lipid peroxidation (as measured by thiobarbituric acid reactive substances or TBARS) occurs; as parallel decreases in antioxidant enzyme activities were observed, it was suggested that conditions of oxidative stress could inactivate CSCitation21. In addition, CS activity was lower in aged compared to young and middle-aged humansCitation22. Finally, a recent studyCitation12 showed that CS activity falls significantly under conditions of oxidative stress (as measured by the ratio of reduced to oxidized glutathione) during cardiac arrest. This, together with evidence that citric acid cycle enzymes may be responsible for the decline in ATP production in the failing heartCitation13, makes a study on CS oxidation and antioxidant protection of CS clinically relevant.
AAPH is a water-soluble and cell-permeable peroxyl radical generator utilized in many studies involving lipid peroxidation and the characterization of novel antioxidantsCitation23,Citation24. Decomposition of AAPH produces nitrogen gas and two carbon-centered radicals, which react with oxygen to produce hydrophilic alkyl peroxyl and alkoxyl radicalsCitation25,Citation26. These radicals differ in their types and rates of reactivity toward proteins from hydroxyl radicals generated during Fe2+/H2O2 (Fenton) reactions. The specific effects of peroxyl radicals on the enzymes of the citric acid cycle remain to be investigated. To our knowledge, ours is the first study to examine the effects of peroxyl radicals on mitochondrial enzymes.
Naphthalenediols (NDs) constitute a novel class of phenolic antioxidants that may be used to restore CS activity, provided that the mechanism of CS inactivation involves free radical attackCitation14,Citation15. NDs are thought to work by transferring a hydrogen atom to either the attacking free radical or to an oxidized amino acid (such as a tyrosyl radical (R-Tyr-O•))Citation27. Our previous studies showed that 1,8-NDs are potent antioxidants with low cytotoxicityCitation14.
Our primary goals were to investigate CS for its susceptibility to oxidation by peroxyl radicals generated by AAPH, both as the purified enzyme and in isolated mitochondria, as well as to evaluate and compare the ability of both natural (ascorbate (ASC), glutathione (GSH), propyl gallate (PG), and epigallocatechin gallate (EGCG)) and synthetic antioxidants (1,4-, 1,8-, and 2,3-naphthalenediols (1,4-ND, 1,8-ND, and 2,3-ND, respectively)) to protect CS against AAPH-induced inactivation.
Materials and methods
Chemistry
1,4-ND and 2,3-ND were obtained from Sigma-Aldrich (Oakville, Ontario). 1,8-ND was synthesized from 1,8-naphthosultone (Sigma-Aldrich) as previously describedCitation28 (for an alternative method of synthesis refer to reference 29). All structures were verified by 1H and 13C nuclear magnetic resonance (NMR) and mass spectrometry. 1,8-ND was a kind gift from Dr. Tony Durst (Chemistry, University of Ottawa, Ottawa, Ontario, Canada).
Materials
Purified porcine heart CS was purchased from Roche Diagnostics (Mannheim, Germany). Reagents for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; Protogel) were from National Diagnostics (Atlanta, Georgia). Prestained protein markers were from New England Biolabs (Beverly, Massachusetts). Chelex-100 and protein dye reagent were purchased from Bio-Rad Laboratories (Hercules, California). All other reagents were purchased from Sigma-Aldrich. Trypsin gold (mass spectrometry grade) was purchased from Promega.
Cell culture, mitochondrial isolation, and treatment with glucose and AAPH
PC12-AC cells, an adherent clone of the rat adrenal pheochromocytoma cell line, were a gift from Dr. Steffany Bennett (Biochemistry, Microbiology and Immunology, University of Ottawa, Ontario, Canada). The cells were thawed and grown in RPMI 1640 medium supplemented with 5% newborn calf serum, 10% heat-inactivated horse serum, glutamine, bicarbonate, 100 U/mL penicillin G (sodium salt), 100 μg/mL streptomycin sulfate, and 0.25 μg/mL amphotericin B (Invitrogen, Carlsbad, California). The cells were maintained at 37°C in a humidified atmosphere (5% CO2 in air) and passed at a density of approximately 5 × 105 cells/mL twice a week.
Mitochondria were isolated from cells as previously describedCitation30. Briefly, cells were detached from plates using a cell scraper into phosphate buffered saline (PBS) and centrifuged at 600g for 10 min at 4°C. Cells were homogenized in IBc buffer without sucrose (10 mM Tris-MOPS, pH 7.4, 1 mM EGTA/Tris) with 40 strokes using a glass–Teflon potter at 1600 r.p.m. The supernatant was collected and centrifuged at 7000g for 10 min at 4°C. The resulting pellet of mitochondria was washed with IBc buffer without sucrose and centrifuged again at 7000g for 10 min at 4°C. Mitochondria were resuspended in a small volume of IBc buffer without sucrose and used for treatment with glucose and AAPH.
Initial studies showed that the CS in isolated mitochondria was susceptible to AAPH inactivation at concentrations similar to that of purified CS (results not shown). Mitochondria were therefore incubated with either 30 mM glucose or 40 mM AAPH, or a combination of both treatments, at 37°C for either 8 or 24 h. Aliquots of treated mitochondria were removed for measurement of CS and aconitase activities or protein determination by Bio-Rad (Hercules, California) protein dye reagent.
Treatment of purified CS with AAPH or EGCG
Purified CS was diluted to final concentrations of 6 or 12 μM (0.29 or 0.58 mg/mL, respectively) in 150 mM potassium phosphate buffer (pH 7.3) prepared by combining appropriate volumes of mono- and dibasic potassium phosphate until the desired pH was achieved. Stock solutions of AAPH were prepared at a concentration of 700 mM in ultra-pure Milli-Q water (to minimize transition metal contamination) pre-heated to 37°C for 5 min. The stock solutions (or Milli-Q water for controls) were then added to CS at 40–100 mM final concentration. The reactions were performed in open tubes in an Isotemp hybridization oven (Fisher Scientific, Ottawa, Ontario), which provided constant air ventilation ensuring sufficient oxygen supply for the AAPH-mediated alkyl peroxyl radical formation. The reactions were stopped by the addition of excess (36-fold) sample dilution buffer (0.1 M Tris-HCl, pH 7.0) and assayed immediately to prevent CS oxidation during the enzyme assay. Dilution in Tris-HCl, a potent free radical scavenger at this concentrationCitation31, and transfer of samples from 37 to 25°C ensured negligible free radical production during the course of enzyme assaysCitation32. Non-interference of residual AAPH or free radicals present in solution with the enzyme assay was verified by the addition of equal amounts of either reaction buffer (control) or assay concentration of AAPH to CS and performing enzymatic assays. For EGCG treatment, the same conditions were used except that the concentration of the EGCG stock (15 mM in 150 mM potassium phosphate buffer, pH 7.3) was prepared immediately prior to the experiments and was kept on ice until addition to CS samples to minimize possible autooxidation.
Enzymatic activity
CS enzymatic activity was measured by monitoring the appearance of thionitrobenzoate at 412 nm (ϵ = 13.6 mM− 1 cm−1) from DTNB (5,5′-dithiobis(2-nitrobenzoic acid)) as a result of its reaction with coenzyme A (CoA-SH), as described previouslyCitation33,Citation34. The assay solution contained 0.25 mM acetyl-CoA, 0.5 mM oxaloacetate (OAA), 0.25% (v/v) Triton X-100, and 0.1 mM DTNB in 100 mM Tris-HCl buffer, pH 8.1. Under these conditions, the initial enzyme velocity was directly proportional to the CS concentration (data not shown). The assays were performed at 25°C and the reaction was initiated by the addition of 10 μL of reaction mix (0.17 μM of porcine CS or 10 μL of isolated mitochondria and varying concentrations of AAPH) to 90 μL of assay mixture (100 μL total reaction volume) pre-incubated at 25°C for 5 min prior to CS addition. A microplate reader (Spectromax 340PC; Molecular Devices, Sunnyvale, California) was used for the assays. Chelex-100 treatment of CS reaction buffer (batch method following the manufacturer’s instructions) prior to AAPH treatment produced results identical to Chelex-100 untreated samples (data not shown), suggesting that contaminating transition metals were not a factor in our experiments. Full (100%) CS activity was defined as the amount of enzyme in the assay that produced 80.4 ± 2.0 μmol of citrate min−1 mg−1 CS (80.4 ± 2.0 IU mg−1 CS). One unit of mitochondrial CS activity was defined as the amount of enzyme in the assay required to convert 1 μmol of OAA and acetyl-CoA to citrate per min.
Aconitase activity was assayed according to Tretter and Adam-ViziCitation10. Briefly, 100 μL of isolated mitochondria was assayed for activity in a buffer containing 50 mM Tris-HCl, pH 7.4, 0.2% Triton X-100, 30 mM sodium citrate, 0.6 mM manganese chloride, 0.2 mM NADP+, 1 U/mL catalase, and 0.2 U/mL isocitrate dehydrogenase at 37°C for 20 min with an initial 5 min incubation period. Reactions were initiated with NADP+ and read at 340 nm with an ultraviolet (UV)-visible spectrophotometer (Cary 100 Bio; Varian, Inc., Palo Alto, California). One unit of mitochondrial aconitase activity was defined as the amount of enzyme in the assay required to convert 1 μmol of citrate to isocitrate per min.
Antioxidant protection
To determine the protective effect of various compounds on AAPH-mediated CS inactivation, each antioxidant was added immediately prior to the addition of AAPH and the reactions were allowed to proceed at 37°C as described above. GSH, ASC, and OAA were dissolved in 150 mM potassium phosphate buffer, pH 7.3. NDs and PG were prepared in DMSO (dimethylsulfoxide) so that the final concentration of DMSO in the assay, in both treated and control samples, was 0.2% (v/v). DMSO did not interfere with the assay at concentrations up to 1% (v/v) (data not shown). The IC50 was defined as the concentration of an antioxidant required to prevent 50% loss of CS enzymatic activity as a result of AAPH treatment.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
After AAPH treatment, protein samples were subjected to SDS-PAGECitation35 to examine possible structural modifications. Protein samples (14 μg) were mixed with 4X loading buffer (40% glycerol, 20% β-mercaptoethanol, 8% SDS, and 0.01% bromophenol blue in 250 mM Tris-HCl buffer, pH 6.8), heated for 3 min at 95°C, and loaded into 12.5% polyacrylamide gels. A Mini-PROTEAN 3 system (Bio-Rad Laboratories Inc., Hercules, California) was used and the gels were run for 1 h at 150 V. Gels were stained with staining solution (0.25% Coomassie Brilliant Blue R, 50% (v/v) methanol, 7.5% (v/v) acetic acid), dried in a GelAir Drying System (Bio-Rad, Hercules, California) and scanned with a CanoScan LIDE 80 scanner (Canon, Mississauga, Ontario). Band densitometry was performed using AlphaEaseFC software, Version 3.1.2 (Alpha Innotech, San Leandro, California).
Dityrosine (diTyr) and tryptophan (Trp) measurements
To further characterize oxidative modifications induced by AAPH, CS samples were analyzed for diTyr formation and intrinsic (inherent, native to the protein) Trp fluorescence using a PerkinElmer LS 50 Spectrofluorimeter (Wellesley, Massachusetts). DiTyr fluorescence was measured with the following parameters: excitation λ: 325 nm, emission λ: 360–560 nm, excitation slit width: 2.5 nm, emission slit width: 4.0 nm, scan speed: 50 nm/min. For Trp fluorescence, the excitation λ was 295 nm, with all other parameters of diTyr measurement. CS samples were diluted five-fold with 0.1 M Tris-HCl, pH 7.0 prior to analysis. Non-interference of residual AAPH with each type of measurement was verified by adding either reaction buffer or the appropriate AAPH concentration to CS and measuring the emission intensity.
Mass spectrometry (MS)
Both control and treated (8 h, 40 mM AAPH) CS samples were spun six times through Ultrafree-0.5 columns (molecular weight cut-off 5 kDa; Millipore) by centrifugation at 12,000g for 30 min at 4°C. Each time, 400 μL of 0.1 M ammonium bicarbonate pH 7.5 was added to approximately 100 μL of CS concentrate. Samples were heated for 5 min at 70°C, treated with dithiothreitol (DTT; 5 mM final concentration), and heated at 56°C for 1 h. After cooling to room temperature and adding iodoacetamide (10 μM final concentration; 1 h treatment in dark), 200 μg of each sample was incubated overnight with 10 μg trypsin at 37°C.
Peptides were desalted using ZipTip μC18 tips (Millipore, Billerica, Massachusetts) according to the manufacturer’s instructions and loaded into glass nanoelectrospray capillaries (Proxeon, Odense, Denmark). Mass analysis was conducted on both the control and treated samples using a QSTAR XL mass spectrometer (Applied Biosystems, Foster City, California) equipped with a manual nanoelectrospray ionization source and operating in time-of-flight (TOF) mode. The resultant spectra were manually analyzed and compared in order to discern which peptides contained dityrosine linkages. Several peptides were also analyzed by MS/MS and identified using the commercially available peptide identification software Mascot (Matrixscience, London, UK).
Protein surface hydrophobicity
To determine whether any other structural changes arise as a result of AAPH treatment, protein hydrophobicity was measured. CS samples (1 μM) were incubated with 100 mM of 8-anilino-1-naphthalenesulfonic acid (ANSA), a protein surface hydrophobicity probeCitation36, for 40 min at 37°C. Excitation and emission λ were 370 and 400–600 nm, respectively, with all other parameters of diTyr measurements. AAPH non-interference was verified by comparing AAPH and reaction buffer addition to untreated CS just prior to ANSA incubation.
Statistics
Data are presented as mean with standard error of the mean from no less than three independent experiments. One-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was used for multiple comparisons unless otherwise specified. Data were considered statistically significant when p < 0.05 unless otherwise specified.
Results
Inactivation of mitochondrial enzymes by AAPH
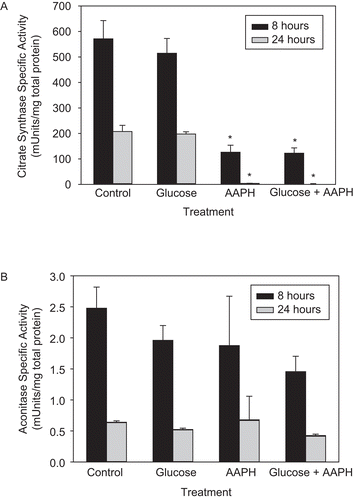
To investigate the effects of treatment of AAPH on CS and aconitase within intact mitochondria, the organelles isolated from PC-12AC were incubated with 40 mM AAPH, and/or with 30 mM glucose to stimulate accelerated cell senescence due to mitochondria-generated oxidative stressCitation25,Citation26. CS and aconitase activities were assayed after 8 and 24 h of treatment (). AAPH had a dramatic effect on CS (), bringing CS activity to 22 and 2% of control rates by 8 and 24 h, respectively. In contrast, aconitase activity did not change significantly with AAPH treatment (). Similar to previous results, we found that H2O2 inactivated aconitase, but not CSCitation10 (and data not shown), in whole mitochondria, suggesting that AAPH- and H2O2-generated free radicals have opposite effects on the activities of these two enzymes.
Figure 1. The effect of 2,2′-azobis(2-amidinopropane) dihydrochloride (AAPH) on the enzymatic activity of mitochondrial citrate synthase (CS) and aconitase. Whole mitochondria were isolated from PC-12AC cells, treated with 30 mM glucose, 40 mM AAPH, or a combination of both for 8 and 24 h at 37°C and assayed for CS and aconitase activities. Ten or 100 μL of the incubation mixture was removed for the assay of CS (A) or aconitase (B) activities, respectively. Means of three independent experiments ± SEM are shown. *Significantly different from untreated controls at p < 0.001 as tested with one-way ANOVA followed by Tukey test.
CS inactivation by AAPH
The susceptibility of CS to AAPH-produced alkyl peroxyl (and alkoxyl) radicals was determined by measuring the loss of its enzymatic activity upon AAPH treatment. As shown in , CS activity was sensitive to the free radicals supplied by thermal decomposition of AAPH. Using the formula Ri = 1.36 × 10−6[AAPH], where Ri is the rate of free radical generation in M/sCitation23, the amount of free radicals required for 50% AAPH-provoked loss of CS activity was found to be 0.34, 0.35, and 0.38 mM for 40, 80, and 100 mM AAPH respectively. This corresponded to 113-, 118-, and 125-fold excess of radicals over CS. Since both substrates for the enzymatic reaction were at saturating concentrations and CS activity was proportional to its concentration, 50% inactivation of the enzyme corresponds to the oxidative damage leading to the loss of activity of half of the total CS used (3 μM). Therefore, one CS molecule is inactivated by 118 ± 6 AAPH-derived radicals. The amount of AAPH required to inactivate CS is much higher than that required to inactivate other enzymes (), revealing relative resistance of CS to oxidative modification. Even though high AAPH concentrations were used, the rate of ROS generation during AAPH decomposition was approximately the same order of magnitude as the maximal rate of mitochondrial ROS generation (0.2 nmol H2O2 min−1 mg−1 proteinCitation43).
Figure 2. (A) CS inactivation by AAPH. CS (6 μM) was incubated with various concentrations of AAPH. Sample aliquots were taken at the time intervals indicated, the reaction was stopped by excessive dilution in 0.1 M Tris-HCl, pH 7.0, and the remaining enzymatic activity was measured immediately, as described in “Materials and methods.” Means of at least three independent experiments ± SEM are shown. (B) CS aggregates as a result of AAPH treatment. CS (12 μM) was incubated with 40 mM AAPH, and aliquots were removed at various time intervals as shown, frozen immediately and stored at −80°C until use. For sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), 14 μg of total protein was loaded per lane and the electrophoresis was run followed by Coomassie staining, as described in “Materials and methods.” Representative gel of three independent experiments is shown. C is control and T is AAPH-treated sample. M and A denote the CS monomer and aggregates, respectively.
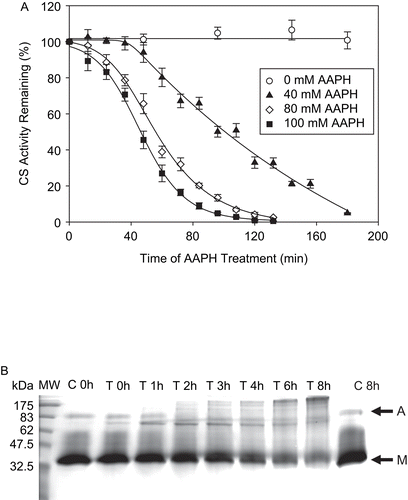
Table 1. The susceptibility of different enzymes to 2,2′-azobis (2-amidinopropane) dihydrochloride (AAPH)-mediated inactivation.
Protein aggregation
In order to further characterize the protein oxidative damage as a result of the pro-oxidant treatment, SDS-PAGE analysis was performed. It allows monitoring gross structural protein modifications such as aggregation and fragmentation. CS migrated with an apparent molecular weight of approximately 39 kDa, while the estimated monomeric molecular weight of CS is 46 kDaCitation44. This error may be due to the use of pre-stained proteins as markers, whose mobility may be altered by dye molecules. reveals that progressively increasing protein aggregation occurs with AAPH treatment over time. At the same time, the intensity of the CS monomer gradually diminishes. The band density of the monomer decreased significantly at 6 and 8 h of treatment compared to controls (p < 0.001; data not shown). In addition, no protein fragments (bands) are seen on the gel below the CS monomer band. The persistence of higher molecular weight species under highly reducing conditions of SDS-PAGE (700 mM β-mercaptoethanol) as well as the failure of 10 mM DTT to restore the CS activity after AAPH treatment (results not shown) rules out the possibility of protein aggregation due to intermolecular disulfide bridge formation.
Increase in protein surface hydrophobicity
Another AAPH-provoked oxidative modification observed was the increase in CS surface hydrophobicity, as measured by the extent of ANSA binding. This method is used to determine the accessibility of protein hydrophobic regions to the hydrophobicity probe ANSA, which can change as a result of protein partial unfolding or increases in protein flexibilityCitation45. reveals that the emission intensity of ANSA binding increases with the time of AAPH treatment, while no change is observed for control CS. The emission intensity increase and the characteristic blue shift observed () correspond to ANSA binding to protein and ANSA transfer from a hydrophilic to a hydrophobic environmentCitation45. The binding shows a biphasic pattern, increasing linearly in the first 120 min of AAPH treatment, after which it remains unchanged (). The increase in ANSA binding may indicate protein unfoldingCitation45 or decreased association between two subunits of the CS dimer, exposing hydrophobic regions in the subunit interfaces that would be otherwise less accessible to ANSA.
Figure 3. AAPH-induced increase in CS surface hydrophobicity. CS (6 μM) was incubated with 40 mM AAPH, aliquots were removed at several time points, diluted, and 1 μM CS was incubated with 100 μM 8-anilino-1-naphthalenesulfonic acid (ANSA) for 40 min at 37°C. The emission intensity was measured upon excitation at 325 nm, as described in “Materials and methods.” (A) Hydrophobicity spectra of AAPH-treated CS over time. (B) Increase in the emission intensity at 490 nm, characteristic for ANSA binding to protein surface hydrophobic residues. Means of at least three independent experiments ± SEM are shown.
Loss of tryptophan fluorescence
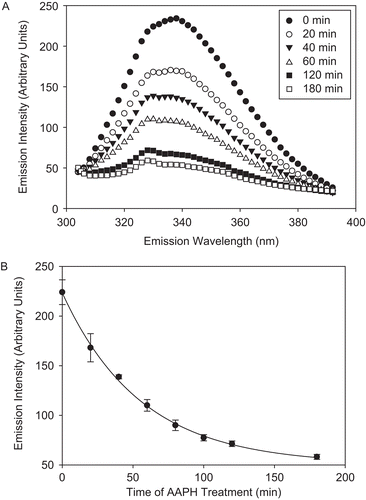
In addition, exposure of CS to AAPH-derived radicals resulted in the loss of intrinsic Trp fluorescence (). Trp fluorescence was observed at λexc of 295 nm, which measures only Trp and not TyrCitation46. The emission spectra of CS had a λmax of 339 nm, suggesting that the overall environment of its nine Trp residues is relatively hydrophobic. Since in a folded protein not all Trps contribute to the fluorescent signal equally (as a result of quenching of their fluorescence by adjacent residues, for example), only the fluorescence of unquenched Trp residues was measured. The AAPH-mediated loss of Trp fluorescence was accompanied by an 11 nm blue shift, indicative of a slight overall decrease in the polarity of oxidized CSCitation47. It is noteworthy that both Trp fluorescence and surface hydrophobicity experiments pointed to an increased hydrophobic environment of AAPH-treated CS.
Figure 4. Kinetics of tryptophan (Trp) fluorescence loss of CS as a result of AAPH treatment. CS (6 μM) was incubated with 40 mM AAPH, aliquots were removed at several time points, diluted, and the fluorescent emission intensity was measured at 300–400 nm upon excitation at 295 nm, as described in “Materials and methods.” (A) Trp fluorescence spectra of AAPH-treated CS over time. (B) Decrease in the emission intensity at 330 nm. Means of three independent experiments ± SEM are shown.
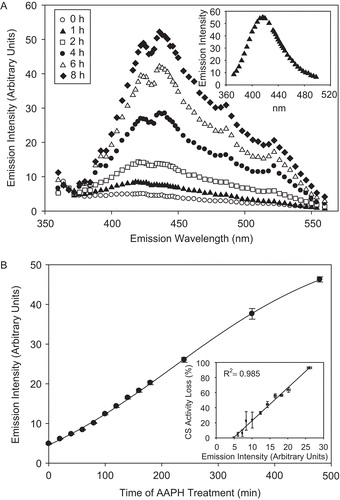
Dityrosine formation
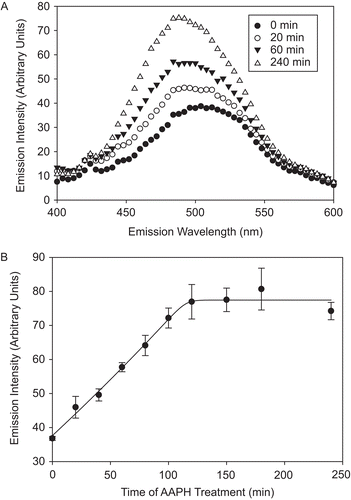
The appearance of a diTyr peak measured at λem of 370–560 nm and λexc of 325Citation38 was also evident upon treatment of CS with AAPH (). The increase in emission intensity at 420 nm, indicative of diTyr formationCitation38, was essentially linear during the first 8 h of AAPH treatment, with a plateau occurring toward the end of the treatment (). The increase in emission intensity at 420 nm using λexc of 325, characteristic of diTyr, correlates well (R2 = 0.98) with AAPH-provoked CS activity loss ( inset), suggesting that Tyr cross-linking may be responsible for the loss of CS enzymatic activity reported in this work.
Figure 5. CS exposure to AAPH leads to dityrosine (diTyr) formation. CS (12 μM) was incubated with 40 mM AAPH, and aliquots were removed and fluorescent emission intensity was measured using excitation wavelength of 325 nm, as described in “Materials and methods.” (A) diTyr spectra of AAPH-treated CS over time. (Inset) The emission intensity of 1 mM Tyr treated with 40 mM AAPH for 2 h at 37°C and excited at 325 nm. (B) Increase in emission intensity at 420 nm, indicative of diTyr formation. (Inset) The correlation between diTyr formation (as measured by emission intensity) and CS activity loss. Means of three independent experiments ± SEM are shown.
Loss of Tyr-containing peptides
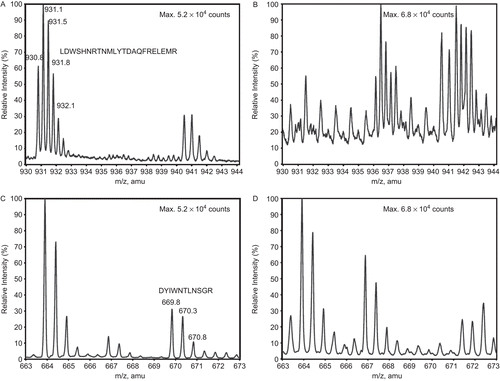
In order to determine whether diTyr was forming as a result of CS treatment with alkyl and alkoxyl radicals, we subjected CS samples to MS analysis. The disappearance of m/z peaks representing Tyr-containing peptides of CS participating in the diTyr formation during AAPH treatment would be expected to occur with the concomitant appearance of diTyr dipeptide peaks. The sequence coverage of CS was 28% at a cut-off of 5000 counts. Interestingly, two out of 14 trypsin-generated peptides that contained Tyr were present in the control, but not in the treated sample. These were LDWSHNRTNMLGYTDAQFRELMR and DYIWNTLNSGR (), containing Tyrs 246 and 331, respectively. Further MS/MS confirmed the sequences of these two peptides, as well as SMSTDGLIK, from the control sample.
Figure 6. The disappearance of CS Tyr-containing peptides during AAPH treatment, as assessed by mass spectrometry. (A) Peptide LDWSHNRTNMLGYTDAQFRELMR in untreated (with m/z (amu) between 930 and 933 (+2)) and (B) its disappearance in AAPH-treated CS. (C) Peptide DYIWNTLNSGR in untreated (with m/z (amu) between 669 and 671 (+2)) and (D) its disappearance in AAPH-treated CS. Note that the peptide ALGVLAQLIWSR, with m/z (amu) between 663 and 665 (+2), remains unaffected by treatment. The peptides in (A) and (C) contain Tyrs 246 and 331, respectively.
Antioxidant protection
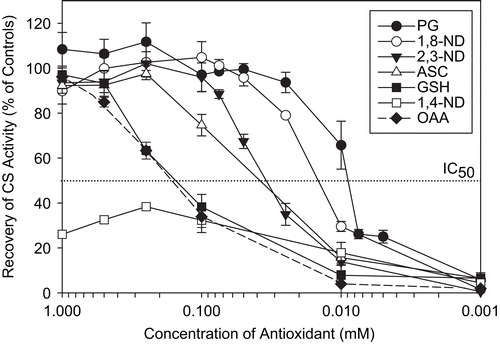
After brief characterization of the types of oxidative damage of CS induced by AAPH, antioxidants shown in were tested for their ability to protect CS against AAPH inactivation. To this end, we added different concentrations of each antioxidant to CS prior to AAPH treatment. Using the results in , we found that the IC50 values for PG, 1,8-ND, 2,3-ND, ASC, GSH, and OAA were 9, 14, 34, 37, 150, and 160 μM, respectively. PG and 1,8-ND offered the best protection, whereas 1,4-ND was unable to protect CS from 50% loss of activity.
Figure 7. Structures of novel antioxidants (1,8-naphthalenediol (ND), 2,3-ND, and 1,4-ND) as well as propyl gallate (PG) and epigallocatechin gallate (EGCG) used to protect CS against inactivation by AAPH.
Figure 8. Protection of CS against AAPH-induced inactivation. CS (6 μM) was incubated with the indicated concentrations of antioxidants prior to 40 mM AAPH treatment for 2 h. CS was then diluted and enzyme assays were performed as described in “Materials and methods.” Untreated CS controls (100% activity), AAPH-treated CS (0% activity), and percentage recovery with respect to AAPH-treated CS are shown. Means of at least three independent experiments ± SEM are presented. The IC50 was taken as the concentration of antioxidant capable of protecting CS against 50% loss of its enzymatic activity as a result of AAPH treatment.
EGCG-mediated inactivation
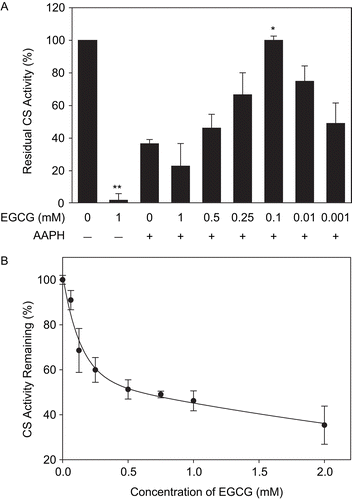
Contrary to our expectations, the antioxidant EGCG was protective against AAPH-mediated loss of CS activity at low concentrations, but inhibitory in an AAPH-additive manner at high concentrations (). EGCG alone was found to induce the loss of CS activity, with approximately 70% inactivation occurring within the first 30 min with 2 mM EGCG ().
Figure 9. EGCG-mediated inactivation of CS and protection against AAPH. (A) CS (6 μM) was incubated with the indicated concentrations of EGCG prior to 40 mM AAPH treatment for 2 h. CS was then diluted and enzyme assays were performed as described in “Materials and methods.” Untreated CS controls (100% activity) and the residual CS activity compared to controls are shown. Means of at least three independent experiments ± SEM are presented. *Significantly different from 40 mM AAPH-treated CS at p < 0.05 as tested with one-way ANOVA followed by Tukey test. **Significantly different from untreated CS controls at p < 0.001 as tested by Student’s t-test. (B) CS (6 μM) was incubated with indicated concentrations of EGCG for 30 min at 37°C, diluted, and the residual activity was measured. Mean values of at least three independent experiments ± SEM are shown.
Discussion
AAPH oxidatively modifies and inactivates CS
Our study has described, for the first time, the susceptibility of CS to oxidative modifications using AAPH as a source of alkyl peroxyl and alkoxyl radicals. AAPH is a slow radical generator, and high concentrations of the compound (40 mM) generate a physiologically relevant flux of free radicals at a constant rateCitation23,Citation24. This concentration of AAPH lowered CS activity (as well as cell viability; EC50 = 10.5 mM for 24 h treatments at a concentration of 4.0 × 105 cells/mLCitation14, and data not shown) in isolated mitochondria and purified CS.
AAPH-generated radicals inactivated CS, but had no effect on mitochondrial aconitase activity (, respectively). In contrast, H2O2 had the opposite effects on these enzymes (data not shown, and reference 10). Purified CS was inactivated by AAPH in a time- and concentration-dependent manner (). Despite the distinguishable induction periods in the CS inactivation curves (), the increase in surface hydrophobicity, Trp loss, and diTyr formation proceeded without any noticeable delay (). This demonstrates that CS can withstand some oxidative damage that does not immediately lead to enzyme inactivation.
CS activity is unchanged by H2O2Citation10, NO•, or O2•–Citation48, which react preferentially with cysteines and methionines, the amino acids most susceptible to oxidationCitation49. H2O2 is a significantly milder oxidative stressor compared to AAPH-derived peroxyl radicals, in the present study, could not inactivate CS (data not shown). Unlike aconitase and α-ketoglutarate dehydrogenase that contain essential cysteinesCitation5,Citation6,Citation8–10 and are inactivated by H2O2, NO•, and O2•–, CS lacking essential Cys residuesCitation50 is resistant to these stressors.
DiTyr, a very common oxidative modification of proteins, has been suggested as a marker of protein oxidationCitation51, and its increased levels are implicated in many pathologiesCitation52. DiTyr forms by the combination of two tyrosyl radicals produced via one-electron oxidation of Tyr. Porcine CS contains 19 Tyr residues (out of 437 residues in total). Given the rather high Tyr content of CS, it is reasonable to expect diTyr formation by the action of alkyl peroxyl radicals in accordance with previous AAPH studiesCitation38,Citation40. Indeed, we observed a significant increase in diTyr emission intensity peak (). Treating a range of Tyr concentrations with 40 mM AAPH over an 8-h period, monitoring the fluorescence emission intensity of diTyr product, and assuming the 100% conversion of Tyr to diTyr, we estimated that approximately 10 (10.3 ± 1.0, n = 3) out of 19 (in total) Tyr residues are converted to five diTyr pairs per CS molecule during 8 h incubation of 12 μM CS with 40 mM AAPH (data not shown).
Close examination of reveals that, in addition to the diTyr emission intensity peak near 420 nm matching that of the diTyr standard ( inset), there are other peaks at approximately 436, 486, and 522 nm. These may represent other fluorescent products such as Trp, kynurenine (λexc 365 nm, λem 480 nm) and N-formylkynurenine (NFK; λexc 325 nm, λem 434 nm)Citation53,Citation54. Since the loss of Trp was observed with AAPH treatment of CS (), the possibility of NFK formation cannot be ruled out based on our results.
The active site of CS consists of residues Arg164, His238, His274, His320, Arg329, Asp375, Arg401, and Arg421, and the adenine recognition loop stretching from amino acids 314 to 320Citation55. OAA binding to the active site initiates the closure of the small domain over the large domain of a single subunit. Domain closure over the substrate is produced by the summation of many small shear motions between pairs of packed helices. The shear motions are facilitated by small deformations in the loops linking the helices. DiTyr formation between two adjacent Tyrs may disrupt binding of substrates or domain closure and have a profound effect on CS activity. Two Tyr–Tyr sequences occur at positions 384,385 and 392,393. DiTyr formation amongst these may affect the shearing motion of the helices during domain closure as they lie at the edge of loop 386–391. Three Tyrs are found close to the active site (Tyrs 318, 330, and 354). Many of the abovementioned Tyrs are within 5 å of each other (190,194; 190,219; 190,392; 194,392; 330,354), and, with opening or closure of the enzyme domains, may be brought even closer for intratyrosine (carbon–carbon) bond formation. The specific Tyr residues involved in diTyr formation in the presence of peroxyl radicals merit further investigation. Mass spectrometry of control and AAPH-treated CS showed that at least two Tyr-containing peptides, LDWSHNRTNMLGYTDAQFRELMR and DYIWNTLNSGR, disappeared in the treated enzyme (). These peptides contain Tyrs 246 and 331, respectively. These Tyrs are found quite distal to one another, both within and between subunits. Peaks were not observed in the treated sample spectrum indicative of either of these two peptides being involved in a diTyr linkage. DiTyr-modified peptides are more difficult to observe by MS relative to non-modified peptides for a number of reasons. The complexity of an AAPH-treated sample would be inherently greater than a control sample, as any Tyr-containing peptide could be involved in at least one diTyr linkage reaction. Increasing sample complexity amplifies the dynamic range and accentuates ion suppression (more competition for available protons), resulting in a relatively lower percentage of peptide species being observed. Moreover, a single peptide could form multiple diTyr linkages with different peptides, lowering its effective concentration. Larger peptides are often more difficult to observe than those of lesser mass; a diTyr-linked peptide would have twice the mass of the average of the individual peptide components.
The possibility that intramolecular disulfide formation led to CS aggregation was ruled out based on the persistence of the aggregates in reducing SDS-PAGE. Similarly, DTT, a strong reducing agent capable of breaking disulfides, failed to restore CS activity (results not shown). Besides diTyr formation, other mechanisms such as carbonyl formation-mediated intermolecular Schiff-base cross-linksCitation56 may be responsible for the CS aggregation observed in this study.
H-atom abstraction from proteins by either ROO• or RO• can destroy some H-bonds crucial for structural integrity, leading to protein unfolding. Hydrophobic residues, which become more exposed to solvent upon protein unfolding, become available for binding to hydrophobic probes such as ANSA; this was probably the case given the increased extent of ANSA binding to CS observed upon AAPH treatment (). The possibility that increased surface hydrophobicity led to CS aggregation is unlikely, as hydrophobic aggregation is typically reversible with SDSCitation57 and, therefore, no higher molecular weight species would be seen on SDS-PAGE. Thus, CS aggregates observed here arise via covalent intramolecular cross-linking.
Antioxidant effectiveness
The overall antioxidant capacity of the compounds tested, in terms of their protective effect against AAPH-mediated CS inactivation, was (from the most to the least protective): PG ≅ 1,8-ND > 2,3-ND > ASC > GSH > OAA > 1,4-ND >> EGCG. The best natural and synthetic antioxidants were PG and 1,8-ND, with IC50 values of 9 and 14 μM, respectively. The antioxidant capacity of synthetic compounds arises from their ability to undergo hydrogen atom abstraction from the phenolic OH groups by peroxyl radicals. This is reflected in their bond dissociation enthalpy (BDE) values, which can be used to predict hydrogen atom transfer feasibility. It is desirable that BDE1 (assigned to the weakest O–H bond) is low, to ensure potent hydrogen atom donation to a free radical. At the same time, the second O–H bond should not be easy to break (high BDE2), in order to avoid quinone formation and toxicity associated with it. The BDEs of 1,8-ND, 2,3-ND, 1,4-ND, and EGCG are BDE1:2 = 72:104, 79:84, 75:55, and 71:74, respectivelyCitation14. Thus, according to BDEs, 1,8-ND should offer the highest protective effects while 1,4-ND should offer the least, which was the case in this study.
Despite its high antioxidant potential and use in the pharmaceutical and food industries (ingredient E-310Citation58), PG can induce cell deathCitation58,Citation59 and DNA fragmentationCitation59. Thus, the NDs were tested as synthetic alternatives to PG. The high antioxidant potential of 1,8-ND seen here is in accordance with our previous studies on rat adrenal pheochromocytoma cellsCitation14 and cortical neuronsCitation15, where 1,8-ND acted as an effective cytoprotector against AAPH-mediated oxidative stress, showing low cytotoxicity. Of the NDs tested, 1,8-ND is the most effective antioxidant due to its ability to donate hydrogen atoms to peroxyl radicals without forming quinones (see references 14, 15, and 27 for an exhaustive overview of antioxidant properties of these phenols). Thus, 1,8-ND provided an effective buffer against AAPH-generated peroxyl radicals, protecting CS activity.
Even though the protective behavior of NDs can be sufficiently explained by their BDE values, the possibility exists that the antioxidant capacity of the compounds stems not from their free radical scavenging properties, but arise as a result of their binding specifically to CS (with dissociation constant (Kd) values as low as 10 μM), perhaps: (1) in the active site of CS, (2) by blocking access of AAPH to its tyrosine targets, or (3) by selecting for protein conformations that do not form diTyr easily. The dissociation constant for the OAA–CS complex is 4.5 ± 1.6 μMCitation60. According to simple calculations assuming chemical equilibrium, CS is 100% saturated with OAA at 250 μM OAA; however, CS was about 30% inactivated at that concentration (). This suggests that the enzyme inactivation observed cannot be entirely attributed to oxidative damage at the OAA active site, such that mechanism (1) as a potential mode of action of the antioxidants is irrelevant, while the possibility of (2) and (3) cannot be ruled out. To account for possible competitive inhibition of CS by NDs, we ran controls with 1 mM of each ND (no AAPH). Only 1,4-ND treatment resulted in about 50% inactivation of CS activity (data not shown).
Antioxidant protection: Other compounds
Significant protection of CS activity was observed with OAA at ≥100 μM (). OAA binding to CS converts CS from its open to its closed form, minimizing the solvent (and hence AAPH in aqueous phase) access to the CS active siteCitation11. Therefore, if AAPH-generated peroxyl radicals oxidize essential residues within the OAA active site, conformational change due to OAA addition may have protected the enzyme against free radical attack.
Full protection of CS activity occurred with GSH concentrations of 0.5–1.0 mM (), an 84-fold excess of GSH over CS. However, the concentration of GSH in mitochondria is approximately one-tenth that of CSCitation61, and therefore GSH is expected to be only weakly protective against peroxyl-induced inactivation and aggregation of CS in vivo.
EGCG-mediated inactivation of CS
To compare the effectiveness of our novel antioxidants against AAPH-induced CS inactivation, EGCG, a known polyphenolic antioxidantCitation62, was used. EGCG has been previously shown to provide pronounced cytoprotection against AAPH-mediated damage, similar to 1,8-NDCitation14,Citation15, in whole cell treatments. In this study, EGCG failed to protect CS against AAPH-dependent inactivation at concentrations of 0.25–1 mM (). Surprisingly, EGCG treatment alone inactivated CS () concentrations several orders of magnitude higher than maximal EGCG blood levels (<1 μM) (see reference 63 and references therein). However, mitochondrial CS concentration is also significantly lower than the 6 μM used here. Therefore, considerably lower EGCG concentrations could, in theory, inactivate CS in vivo.
There are several reasons why EGCG may be cytoprotective in vivo and inhibitory in vitro. First, EGCG is capable of auto-oxidation at neutral pH, producing H2O2 and O2•–Citation64 that could inactivate CS via covalent modifications. However, treatment of CS with bolus addition of H2O2 and O2•– had no effect on CS activityCitation38 (and results not shown). Similarly, H2O2 generation alone cannot be entirely responsible for the pro-oxidant effects of EGCGCitation65,Citation66. Second, EGCG contains ortho-OH groups that can auto-oxidize to very unstable ortho-quinones that easily undergo nucleophilic attackCitation64. Protein nucleophiles react with ortho-quinones via 1-4 Michael addition. However, EGCG quinone is a poor electrophile due to its low pKa (4.3), and thus nucleophilic adducts of EGCG-produced quinones with CS are unlikely. Finally, EGCG, being a water-soluble tanninCitation67, can cross-link proteins reversibly via hydrogen bondsCitation57 or irreversibly via protein–semiquinone radical covalent bond formationCitation68. This mechanism (EGCG-catalyzed protein cross-link formation) appears to be the most probable for the CS inactivation. Supporting this view, EGCG with two phenolic moieties suitable for protein multiple linkagesCitation68, but not PG with only one phenolic moiety, inactivated CS ( and 8).
Use of CS as mitochondrial marker may be misleading
It is currently thought that a decrease in CS activity following oxidative stress during hypoxia–ischemia in the brain occurs as a result of mitochondrial membrane rupture and CS release from mitochondriaCitation69. At the same time, the activity loss of another citric acid cycle enzyme, aconitase, is used as an assessment of oxidative damageCitation69. Many studies use Fe2+/H2O2 as an oxidative stressor and therefore do not provide a full assessment of the effects of different types of radicals on mitochondrial enzyme activities. The present study shows that CS, rather than aconitase, is susceptible to peroxyl radical inactivation. Similarly, in vivo, the activity of CS fell to only 19% of control activity under oxidative stress conditions accompanying cardiac arrestCitation12. Therefore, even though further evidence from animals or tissues subjected to conditions of oxidative stress is required to support this claim, our results strongly suggest that the current practice of using CS as an index of mitochondrial integrity or abundance and the use of aconitase as an oxidative stress marker can lead to erroneous conclusions, especially in studies where oxidative stress is a factor. Decreased CS activity may reflect oxidative inactivation of CS rather than lowered intact mitochondria content.
Acknowledgments
We are grateful to Wendy Bennitz for her editorial comments.
Declaration of interest: This work was supported by a Discovery Grant from the Natural Sciences and Engineering Research Council (NSERC) of Canada to W. G. Willmore, an NSERC Canada Graduate Scholarship (CGS) and Ontario Graduate Scholarship (OGS) to N. L. Chepelev, and an NSERC Undergraduate Student Research Award (USRA) to J. D. Bennitz.
Related Research Data
References
- Lenaz G, Baracca A, Fato R, Genova ML, Solaini G.New insights into structure and function of mitochondria and their role in aging and disease. Antioxid Redox Signal 2006;8:417–37.
- Turrens JF. Superoxide production by the mitochondrial respiratory chain. Biosci Rep 1997;17:3–8.
- Tahara EB, Barros MH, Oliveira GA, Netto LE, Kowaltowski AJ. Dihydrolipoyl dehydrogenase as a source of reactive oxygen species inhibited by caloric restriction and involved in Saccharomyces cerevisiae aging. FASEB J 2007;21:274–83.
- Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, et al. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci 2004;24:7779–88.
- Tretter L, Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos Trans R Soc Lond B Biol Sci 2005;360:2335–45.
- Adam-Vizi V. Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non-electron transport chain sources. Antioxid Redox Signal 2005;7:1140–9.
- Lenaz G. The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life 2001;52:159–64.
- Bulteau AL, Ikeda-Saito M, Szweda LI. Redox-dependent modulation of aconitase activity in intact mitochondria. Biochemistry 2003;42:14846–55.
- Sadek HA, Humphries KM, Szweda PA, Szweda LI. Selective inactivation of redox-sensitive mitochondrial enzymes during cardiac reperfusion. Arch Biochem Biophys 2002;406:222–8.
- Tretter L, Adam-Vizi V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: a key role of α-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J Neurosci 2000;20:8972–79.
- Remington SJ. Structure and mechanism of citrate synthase. Curr Top Cell Regul 1992;33:209–29.
- Sharma AB, Sun J, Howard LL, Williams AG Jr, Mallet RT. Oxidative stress reversibly inactivates myocardial enzymes during cardiac arrest. Am J Physiol Heart Circ Physiol 2007;292:H198–206.
- Sheeran FL, Pepe S. Energy deficiency in the failing heart: linking increased reactive oxygen species and disruption of oxidative phosphorylation rate. Biochim Biophys Acta 2006;1757:543–52.
- Flueraru M, Chichirau A, Chepelev LL, Willmore WG, Durst T, Charron M, et al. Cytotoxicity and cytoprotective activity in naphthalenediols depends on their tendency to form naphthoquinones. Free Radic Biol Med 2005;39:1368–77.
- Flueraru M, So R, Willmore WG, Poulter MO, Durst T, Charron M, et al. Cytotoxicity and cytoprotective activity of naphthalenediols in rat cortical neurons. Chem Res Toxicol 2006;19:1221–7.
- Reisch AS, Elpeleg O. Biochemical assays for mitochondrial activity: assays of TCA cycle enzymes and PDHc. Methods Cell Biol 2007;80:199–222.
- Marin-Garcia J, Ananthakrishnan R, Goldenthal M. Human mitochondrial function during cardiac growth and development. Mol Cell Biochem 1998;179:21–6.
- Drahota Z, Milerova M, Stieglerova A, Houstek J, Ostadal B. Developmental changes of cytochrome c oxidase and citrate synthase in rat heart homogenate. Physiol Res 2004;53:119–22.
- Navarro A, Sanchez Del Pino MJ, Gomez C, Peralta JL, Boveris A. Behavioral dysfunction, brain oxidative stress, and impaired mitochondrial electron transfer in aging mice. Am J Physiol Regul Integr Comp Physiol 2002;282:R985–92.
- Vitorica J, Cano J, Satrustegui J, Machado A. Comparison between developmental and senescent changes in enzyme activities linked to energy metabolism in rat heart. Mech Ageing Dev 1981;16:105–16.
- Freitas JJ, Pompeia C, Miyasaka CK, Curi RJ. Walker-256 tumor growth causes oxidative stress in rat brain. J Neurochem 2001;77:655–63.
- Rooyackers OE, Adey DB, Ades PA, Nair KS. Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc Natl Acad Sci USA 1996;93:15364–9.
- Niki E. Free radical initiators as source of water- or lipid-soluble peroxyl radicals. Methods Enzymol 1990;186:100–8.
- Terao K, Niki E. Damage to biological tissues induced by radical initiator 2,2’-azobis(2-amidinopropane) dihydrochloride and its inhibition by chain-breaking antioxidants. Free Radic Biol Med 1986;2:193–201.
- Książek K, Bręborowicz A, Jörres A, Witowski J. Oxidative stress contributes to accelerated development of the senescent phenotype in human peritoneal mesothelial cells exposed to high glucose. Free Radic Biol Med 2007;42:636–41.
- Książek K, Passos JF, Olijslagers S, von Zglinicki T. Mitochondrial dysfunction is a possible cause of accelerated senescence of mesothelial cells exposed to high glucose. Biochem Biophys Res Commun 2008;366:793–9.
- Hussain HH, Babic G, Durst T, Wright JS, Flueraru M, Chichirau A, et al. Development of novel antioxidants: design, synthesis, and reactivity. J Org Chem 2003;68:7023–32.
- Ragot JP, Steeneck C, Alcaraz M-L, Taylor RJK. The synthesis of 1,8-dihydroxynaphthalene-derived natural products: palmarumycin CP1, palmarumycin CP2, palmarumycin C11, CJ-12,371, deoxypreussomerin A and novel analogs. Perkin Trans 1 1999;8:1073–82.
- Foti MC, Johnson ER, Vinqvist MR, Wright JS, Barclay LRC, Ingold KU. Naphthalene diols: a new class of antioxidants. Intramolecular hydrogen bonding in catechols, naphthalene diols and their aryloxyl radicals. J Org Chem 2002;67:5190–6.
- Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc 2007;2:287–95.
- Searle AJF, Tomasi A. Hydroxyl free radical production in iron-cysteine solutions and protection by zinc. Inorg Biochem 1982;17:161–6.
- Videla LA, Salim-Hanna M, Lissi EA. Inactivation of yeast alcohol dehydrogenase by alkylperoxyl radicals. Characteristics and influence of nicotinamide-adenine dinucleotides. Biochem Pharmacol 1992;44:1443–52.
- Srere PA. Citrate synthase. Methods Enzymol 1969;13:3–11.
- Laboratory protocol: citrate synthase. Oroboros Instruments, Innsbruck, Austria, 2006 April 10. Available at: http://www.oroboros.at/, accessed 2007 Sep 12.
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;227:680–5.
- Weber G, Young LB. Fragmentation of bovine serum albumin by pepsin. I. The origin of the acid expansion of the albumin molecule. J Biol Chem 1964;239:1415–23.
- Jiménez I, Lissi EA, Speisky H. Free-radical-induced inactivation of lysozyme and carbonyl residue generation in protein are not necessarily associated. Arch Biochem Biophys 2000;381:247–52.
- Viner RI, Krainev AG, Williams TD, Schoneich C, Bigelow DJ. Identification of oxidation-sensitive peptides within the cytoplasmic domain of the sarcoplasmic reticulum Ca2+-ATPase. Biochemistry 1997;36:7706–16.
- Lissi EA, Salim-Hanna M, Faure M, Videla LA. 2,2′-Azo-bis-amidinopropane as a radical source for lipid peroxidation and enzyme inactivation studies. Xenobiotica 1991;221:995–1.
- Chao CC, Ma YS, Stadtman ER. Modification of protein surface hydrophobicity and methionine oxidation by oxidative systems. Proc Natl Acad Sci USA 1997;94:2969–74.
- Kang JH, Kim KS, Choi SY, Kwon HY, Won MH, Kang T-C. Protective effects of carnosine, homocarnosine and anserine against peroxyl radical-mediated Cu,Zn-superoxide dismutase modification. Biochim Biophys Acta 2002;1570:89–96.
- Mayo JC, Tan DX, Sainz RM, Lopez-Burillo S, Reiter RJ. Oxidative damage to catalase induced by peroxyl radicals: functional protection by melatonin and other antioxidants. Free Radic Res 2003;37:543–53.
- Batandier C, Guigas B, Detaille D, El-Mir M-Y, Fontaine E, Rigoulet M, et al. The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J Bioenerg Biomembr 2006;38:33–42.
- McEvily AJ, Harrison JH. Subunit equilibria of porcine heart citrate synthase. Effects of enzyme concentration, pH, and substrates. J Biol Chem 1986;261:2593–8.
- Friguet B, Szweda LI, Stadtman ER. Susceptibility of glucose-6-phosphate dehydrogenase modified by 4-hydroxy-2-nonenal and metal-catalyzed oxidation to proteolysis by the multicatalytic protease. Arch Biochem Biophys 1994;311:168–73.
- Ruan K, Li J, Liang R, Xu C, Yu Y, Lange R, et al. A rare protein fluorescence behavior where the emission is dominated by tyrosine: case of the 33-kDa protein from spinach photosystem II. Biochem Biophys Res Commun 2002;293:593–7.
- Zaidi A, Leclere-L’Hostis E, Marden MC, Poyart C, Leclerc L. Heme as an optical probe for studying the interactions between calmodulin and the Ca2+-ATPase of the human erythrocyte membrane. Biochim Biophys Acta 1995;1236:114–18.
- Andersson U, Leighton B, Young ME, Blomstrand E, Newsholme EA. Inactivation of aconitase and oxoglutarate dehydrogenase in skeletal muscle in vitro by superoxide anions and/or nitric oxide. Biochem Biophys Res Commun 1998;249:512–16.
- Shacter E. Quantification and significance of protein oxidation in biological samples. Drug Metab Rev 2000;32:307–26.
- Srere PA. The sulfhydryl groups of citrate-condensing enzyme. Biochem Biophys Res Commun 1965;18:87–91.
- Giulivi C, Davies KJ. Dityrosine: a marker for oxidatively modified proteins and selective proteolysis. Methods Enzymol 1994;233:363–71.
- DiMarco T, Giulivi C. Current analytical methods for the detection of dityrosine, a biomarker of oxidative stress, in biological samples. Mass Spectrom Rev 2007;26:108–20.
- Fukunaga Y, Katsuragi Y, Izumi T, Sakiyama F. Fluorescence characteristics of kynurenine and N’-formylkynurenine. Their use as reporters of the environment of tryptophan 62 in hen egg-white lysozyme. J Biochem 1982;92:129–41.
- Zhang H, Andrekopoulos C, Joseph J, Crow J, Kalyanaraman B. The carbonate radical anion-induced covalent aggregation of human copper, zinc superoxide dismutase and α-synuclein intermediacy of tryptophan- and tyrosine-derived oxidation products. Free Radic Biol Med 2004;36:1355–65.
- Wiegand G, Remington SJ. Citrate synthase: structure, control, and mechanism. Annu Rev Biophys Biophys Chem 1986;15:97–117.
- Stadtman ER. Protein oxidation and aging. Free Radic Res 2006;40:1250–8.
- Hagerman AE, Rice ME, Ritchard NT. Mechanisms of protein precipitation for two tannins, pentagalloyl glucose and epicatechin16 (4→8) catechin (procyanidin). J Agric Food Chem 1998;46:2590–5.
- Fiuza SM, Gomes C, Teixeira LJ, Girão da Cruz MT, Cordeiro MNDS, Milhazes N, et al. Phenolic acid derivatives with potential anticancer properties—a structure-activity relationship study. Part 1: methyl, propyl and octyl esters of caffeic and gallic acids. Bioorg Med Chem 2004;12:3581–9.
- Nakagawa Y, Moldeus P, Moore G. Propyl gallate-induced DNA fragmentation in isolated rat hepatocytes. Arch Toxicol 1997;72:33–7.
- Johnson JK, Srivastava DK. Interaction of ligands with pig heart citrate synthase: conformational changes and catalysis. Arch Biochem Biophys 1991;287:250–6.
- Ghosh S, Pulinilkunnil T, Yuen G, Kewalramani G, An D, Qi D, et al. Cardiomyocyte apoptosis induced by short-term diabetes requires mitochondrial GSH depletion. Am J Physiol Heart Circ Physiol 2005;289:H768–76.
- Hu C, Kitts DD. Evaluation of antioxidant activity of epigallocatechin gallate in biphasic model systems in vitro. Mol Cell Biochem 2001;218:147–55.
- Lee M-J, Wang Z-Y, Li H, Chen L, Sun Y, Gobbo S, et al. Analysis of plasma and urinary tea polyphenols in human subjects. Cancer Epidemiol Biomarkers Prev 1995;4:393–9.
- Roginsky V, Alegria AE. Oxidation of tea extracts and tea catechins by molecular oxygen. J Agric Food Chem 2005;53:4529–35.
- Elbling L, Weiss RM, Teufelhofer O, Uhl M, Knasmueller S, Schulte-Hermann R, et al. Green tea extract and (-)-epigallocatechin-3-gallate, the major tea catechin, exert oxidant but lack antioxidant activities. FASEB J 2005;19:807–9.
- Dashwood WM, Orner GA, Dashwood RH. Inhibition of beta-catenin/Tcf activity by white tea, green tea, and epigallocatechin-3-gallate (EGCG): minor contribution of H2O2 at physiologically relevant EGCG concentrations. Biochem Biophys Res Commun 2002;296:584–8.
- Wroblewski K, Muhandiram R, Chakrabartty A, Bennick A. The molecular interaction of human salivary histatins with polyphenolic compounds. Eur J Biochem 2001;268:4384–97.
- Hagerman AE, Roger TD, Davies MJ. Radical chemistry of epigallocatechin gallate and its relevance to protein damage. Arch Biochem Biophys 2003;414:115–20.
- Sutherland BA, Shaw OM, Clarkson AN, Jackson DM, Sammut IA, Appleton I. Neuroprotective effects of (-)-epigallocatechin gallate following hypoxia-ischemia-induced brain damage: novel mechanisms of action. FASEB J 2005;19:258–60.