Abstract
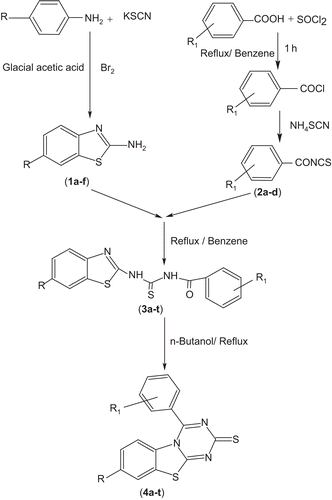
A number of new 8-substituted-4-(2/4-substituted phenyl)-2H-[1,3,5]triazino[2,1-b][1,3]benzothiazole-2-thiones (4a–t) were synthesized and evaluated for their anticonvulsant, anti-nociceptive, hepatotoxic, and neurotoxic properties. The titled compounds (4a–t) were obtained by cyclization of N-{[6-substituted-1,3-benzothiazol-2-yl)amino]carbonothioyl}-2/4-substituted benzamides (3a–t) by refluxing in n-butanol. All the newly synthesized compounds were screened for their anticonvulsant activity in a mouse seizure model and were compared with the standard drug phenytoin. Compounds 4a, 4c, 4f, and 4l showed complete protection after time periods of 0.5 h and 4 h. Some of the selected compounds were evaluated for their neurotoxic and hepatotoxic effects, and none of these showed any sign of neurotoxicity or hepatotoxicity. Compounds 4a–t were also evaluated for their anti-nociceptive activity by a thermal stimulus technique using diclofenac as standard. Compounds 4o, 4q, and 4t displayed highly potent analgesic activity with p < 0.01.
Introduction
Among the different neurological disorders that affect the human condition, epilepsy has been extensively studied during the last century, becoming a dynamic research field in recent years. It describes disorders characterized by recurrent seizure attack due to synchronous neuronal firing. Seizures remain uncontrolled in at least 30% of all epilepsies, even when adequate antiepileptic drug therapy is administered. The classical antiepileptic drugs comprise phenobarbital, available since 1911; phenytoin, marketed in 1939; carbamazepine, used for epilepsy in Europe from the mid-1960s; and valproic acidCitation1, available in several European countries since the late 1960s. All currently approved antiepileptic drugs have dose-related toxicity and idiosyncratic side effectsCitation2. In recent years, benzothiazole derivatives have gained conspicuous significance due to their wide spectrum of biological activities. Although they have been known for a long time to be biologically activeCitation3–5, their varied biological features remain of great scientific interest. Newer anticonvulsant agents having a differently substituted benzothiazole nucleus have been reported to possess significant activitiesCitation6–8.
In our previous work we have reportedCitation9–13 several benzfused five-membered heterocyclic compounds, including the benzothiazole moiety, which have shown marked anticonvulsant activity. In continuation of our research on the benzothiazole moiety, we have combined the benzothiazole pharmacophore with the active triazine pharmacophore and studied the effect on anticonvulsant activity thereafter.
Analgesia is a state of reduced awareness to pain, and analgesics are substances that decrease pain sensation by increasing the threshold to painful stimuli. The management of analgesic disorders involves a stepwise approach to the use of therapeutic agents. Relief of pain and reduction of inflammation are major goals in reducing the severity of symptomsCitation14. A generally accepted stepwise approach to manage painful (nociceptive) disorders includes physical therapy, non-steroidal anti-inflammatory drugs (NSAIDs), disease modifying anti-rheumatic drugs (DMARDs), corticosteroids, and immunosuppressive agents. In recent years, a number of benzothiazole derivatives have been synthesized and found to display analgesic and anti-inflammatory activityCitation15,Citation16.
In the present investigation, we have synthesized 8- substituted -4-(2/4-substituted phenyl)-2H-[1,3,5] triazino [2,1-b][1,3]benzothiazole-2-thiones (). The compounds were evaluated in vivo for anticonvulsant activity by the maximal electroshock seizure (MES) test, anti- nociceptive activity by the thermal stimulus technique, and neurotoxicity by the rotarod method.
Scheme 1. Synthetic route of titled compounds 4a–t.
Materials and methods
Animals
Albino mice (Swiss, 25–30 g) were used in groups of six each as experimental animals. All the test compounds and standard drug were suspended in polyethylene glycol (PEG) and administered intraperitoneally. The animals were maintained on an adequate diet and allowed free access to food and water except during the short time they were removed from their cages for testing. The animals were maintained at room temperature (25 ± 2°C). All experimental protocols were carried out with permission of the Institutional Animal Ethics Committee (IAEC). Animals were obtained from the Central Animal House Facility, Jamia Hamdard University, New Delhi (registration number and date of registration of the Animal House Facility: 173/CPCSEA, 28 Jan 2000).
Chemistry
Solvents selected were of LR (laboratory reagent) grade and were obtained from Merck, CDH, and s.d. Fine Chemicals. Melting points were determined in open capillary tubes and are uncorrected. Thin layer chromatography was performed on silica gel G (Merck). Spots were developed in an iodine chamber and visualized under ultraviolet (UV) light. Infrared (IR) spectra were recorded in KBr pellets on a Bio-Rad FTS 135 WIN-IR spectrophotometer. 1H nuclear magnetic resonsance (NMR) spectra were recorded on a Bruker model DPX 300 FT NMR spectrometer in DMSO-d6 using tetramethylsilane (Me4Si; TMS) as internal standard. The chemical shifts (δ) are recorded in ppm.
General procedure for the synthesis of titled compounds 4a–t (Scheme 1)
6-Substituted-1,3-benzothiazol-2-amines (1a–f) Substituted anilines (0.01 mol) and potassium thiocyanate (0.01 mol) were dissolved in glacial acetic acid, cooled, and stirred for 15 min. Cold bromine solution (0.01 mol, 3 mL in 10 mL acetic acid) was added dropwise. Stirring was continued for an additional 3 h. The separated hydrochloride salt was filtered off, washed with acetic acid, dissolved in hot water, and neutralized with aqueous ammonia solution (25%). The resulting precipitate was filtered off, washed with water, and recrystallized from ethanol to get the desired compounds 1a–f.
2/4-Substituted benzoylisothiocyanates (2a–d) Substituted benzoic acid (0.1 mol) and thionyl chloride (0.1 mol) were refluxed in benzene (50 mL) (CARE—carcinogenic) for 1 h. The reaction mixture upon filtration yielded substituted benzoyl chloride as a viscous liquid. Ammonium thiocyanate (0.1 mol) was added to the substituted benzoyl chloride (0.1 mol) and refluxed for 30 min in benzene. The resulting mixture was filtered and the substituted benzoyl isothiocyanates 2a–d were obtained in the form of a liquid with ammonium chloride as a solid residue. Ammonium chloride was removed by filtration as a residue from the mixture.
N-{[(6-substituted-1,3-benzothiazole-2-yl)amino]carbonothioyl}-2/4-benzamides (3a–t) The compounds 3a–t were obtained when 6-substituted benzothiazoles (1a–f, 0.02 mol) and substituted benzoyl isothiocyanates (2a–d, 0.02 mol) were refluxed in benzene for 5 h. The solid material obtained was filtered and recrystallized from benzene.
8-Substituted 4-(2/4-substituted phenyl)-2H-[1,3,5] triazino [2,1-b][1,3]benzothiazole-2-thiones (4a–t) A solution of compounds 3a–t (0.01 mol) in n-butanol (30 mL) was refluxed for 5 h to obtain compounds 4a–t, which were crystallized from n-butanol. The spectral data and physicochemical properties of compounds 4a–t are given in and .
Table 1. Spectral characterization of compounds 4a–t.
Table 2. Physicochemical properties of compounds 4a–t.
Pharmacology
Anticonvulsant activity
Maximal electroshock seizure test (MES) Initial anticonvulsant evaluation of the test compounds was undertaken by following the anticonvulsant drug development (ADD) program protocolCitation17,Citation18 devised by the Epilepsy Section of the National Institute of Neurological Disorders and Stroke (NINDS), National Institutes of Health (NIH), USA. Each compound was administered as an intraperitoneal (i.p.) injection at a dose level of 30 mg/kg body weight, and the anticonvulsant activity was assessed 0.5 h and 4 h after administration. Maximal electroshock seizures were elicited in mice by delivering 60 Hz, 50 mA electrical stimuli for 0.2 s via ear-clip electrodes. The maximal seizure typically consisted of a short period of tonic extension of the hind limbs and a final clonic episode. Blockade of the hind limbs’ tonic extensor component due to drug treatment was taken as the end point.
Anti-nociceptive activity
Thermal stimulus technique The anti-nociceptive activity was evaluated using a previously described procedureCitation19. The test compounds were suspended in a methylcellulose–water (0.5%) mixture. Each compound was administered orally at a dose of 20 mg/kg. The anti-nociceptive activity was assessed 4 h after administration. The tail of each mouse was gently immersed in thermostatically controlled water at 55°C. The parameter measured in test samples was the time that elapsed between immersion and the attempt to withdraw the tail from hot water, for control as well as treated groups of animals.
Neurotoxic effects
Rotarod test Minimal motor impairment was measured in mice by the rotarod testCitation20. The mice were trained to stay on an accelerating rotarod that rotated at 10 rev/min. The rod diameter was 3.2 cm. Trained animals were given i.p. injections of the test compounds at a dose of 30 mg/kg. Unimpaired mice can easily remain on a rod rotating at this speed. Neurotoxicity was indicated by the inability of the animal to maintain equilibrium on the rod for at least 1 min in each of three concurrent trials.
Histopathological studies
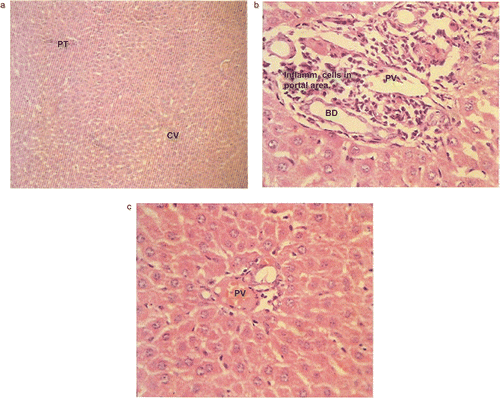
The cellular effects of selected compounds 4a and 4o were evaluated by histopathology. Luna’s techniqueCitation21 was used to assess the livers of the mice, which had received the test compounds at the dose of 30 mg/kg body weight for 15 days. Comparison was made with the control group receiving the dosing vehicle. Microphotographs of liver sections were taken at magnifications of × 100 and × 400.
Log P determination
The desired log P value depends on the nature of the compounds and the testing system. A log P of approximately 2.0 is considered to be the best predictor for penetration of the blood–brain barrier and therefore of central nervous system (CNS) activityCitation22. In this study, we attempted to correlate the anticonvulsant activity with 100% protection against seizure spread in the anti-MES screen with calculated log P values (CLOGP). Log P values were determined for compounds 4a, 4c, 4f, 4g, 4l, and 4m. Experimental log P values were determined using the octanol–water methodCitation23 and CLOGP values were calculated using ACD freeware version 7.1.
Estimation of liver enzymes and proteins
Serum glutamate oxaloacetate transaminase (SGOT) This is a mitochondrial enzyme present in large quantities in the liver, heart, skeletal muscles, and kidneys. It is released from the cells when the tissues are damaged. It was estimated using Rietman and Frankel’s methodCitation24–26.
Serum glutamate pyruvate transaminase (SGPT) This cytosolic enzyme is present abundantly in liver cells. Thus, the serum levels of SGPT are elevated by liver disease. This is considered one of the most sensitive indications of liver damage, particularly in viral hepatic necrosis, e.g. viral hepatitis or toxin-induced liver injury. It was determined using Rietman and Frankel’s methodCitation24–26.
Alkaline phosphatase Alkaline phosphatases are enzy mes that catalyze removal of the phosphate group from monophosphate esters under alkaline conditions. This reaction is of considerable importance in several liver diseasesCitation27.
Estimation of proteins Determination of the proteins provides most useful information in chronic liver diseases. They were determined using the Biuret methodCitation28,Citation29.
Statistical analyses
All statistical analyses were carried out using SigmaStat 4.0 software by means of analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test, and the results are expressed as mean ± SEM.
Results and discussion
Anticonvulsant activity
The anticonvulsant activity and neurotoxicity data for the compounds are reported in . At the dose level of 30 mg/kg, all compounds exhibited average to good protection. Results are presented as percent protection at time intervals of 0.5 h and 4 h. The anticonvulsant drug phenytoin was used as the standard at a dose of 30 mg/kg.
Table 3. Anticonvulsant, anti-nociceptive, and neurotoxicity data of compounds 4a–t.
Compounds 4a, 4c, 4f, 4g, 4l, and 4m showed 100% protection after the 0.5 h time period and the compounds 4g and 4m showed 83% protection after 4 h. All the compounds, with the exception of compound 4s (which showed 50% protection), showed 66–83% protection at both time intervals.
Neurotoxicity screening of compounds is also presented in . In the neurotoxicity screen all the selected compounds were devoid of neurotoxicity at the dose of 30 mg/kg body weight.
Anti-nociceptive activity
8-Substituted-4-(2/4-substituted phenyl)-2H-[1,3,5]triazino [2,1-b][1,3]benzothiazole-2-thiones (4a–t) were evaluated for their anti-nociceptive activity by the thermal stimulus technique. Results of the screening are presented in . Results are reported as mean average reaction time in seconds for untreated control and treatment compounds at the dose level of 20 mg/kg. Diclofenac was used as the standard for comparison. The level of significance was determined by Student’s t test. Compounds 4o, 4q, and 4t showed highly potent activity with p < 0.001, whereas compounds 4a, 4b, 4c, 4d, 4h, 4j and 4s showed significant anti-nociceptive activity with p < 0.01. Compounds 4e, 4i, 4l and 4p also displayed the activity with p < 0.05, however compounds 4f, 4g, 4k, 4m, 4n, and 4r were devoid of anti-nociceptive activity at the tested dose.
Hepatotoxicity studies
Microphotographs of sections of the livers of mice receiving compounds 4a and 4o along with control are presented in . Liver samples from control group animals and all experimental groups were within normal histological limits, except sample 4a which showed moderate portal inflammation. These changes were non-specific in nature. No hepatocyte necrosis or degeneration was seen in any of the samples.
Figure 1. (a) Low power (HE, × 100) and (b, c) high power (HE, × 400) photomicrographs of liver. Sample from control (a) shows normal hepatic parenchyma with portal triad (PT), central vein (CV), and hepatocytes. In the case of compound 4a (b), liver shows hepatic parenchyma with inflammatory cells infiltrating the portal triad (PT). The central vein and hepatocytes do not show any inflammatory cells. BD, Biliary duct. In the case of compound 4o (c), liver shows normal portal triad structures.
Log P determination
Experimental log P values of compounds 4a, 4c, 4f, 4g, 4l, and 4m were found to be 2.80 ± 0.24, 2.78 ± 0.35, 3.10 ± 0.47, 3.12 ± 0.21, 3.16 ± 0.54, and 3.17 ± 0.13, respectively, while their calculated values were 3.46 ± 0.86, 1.52 ± 1.08, 4.44 ± 0.90, 4.26 ± 0.87, 4.23 ± 0.87, and 3.69 ± 0.90, respectively. All the selected compounds were lipophilic in nature.
Estimation of liver enzymes and proteins
Enzyme estimation was performed for the most active compounds (4a and 4o) and the data are presented in . All enzymes estimated were compared to the control. Alkaline phosphatase and SGPT values were found to be increased slightly by both compounds (p < 0.01). Compound 4a also increased the SGOT level in blood slightly (p < 0.01).
Table 4. Effect of selected compounds on enzyme levels.
The two proteins found in the liver, albumin and globulin, were also estimated, and the albumin/globulin ratio was also determined. The results are presented in . All parameters determined were compared to the control. In advanced liver disease, albumin decreases and globulin often increases. It was found that the albumin level in blood increased significantly after administration of the two compounds. There was no significant change in globulin level.
Table 5. Effect of selected compounds on protein levels.
Conclusions
All the compounds were active in the MES test, indicative of their ability to prevent seizure spread. Maximum potency was observed when halogen substituents such as Br, Cl, and F and nitro substituent at the 8-position of the benzothiazole ring with a 2/4-Cl substituted phenyl ring were present in the compounds. However, substitution with CH3 at the 8-position of the benzothiazole ring with 4-OCH3 at the phenyl ring reduced the potency by 50%. Derivatives with OCH3 at the 8-position of the benzothiazole ring and 4-Cl/OCH3 substituent at the phenyl ring exhibited a decrease in activity to a lesser extent.
The anti-nociceptive activity of the compounds was evaluated by a thermal stimulus technique. Substitution with 8-F, 8-OCH3 at the benzothiazole ring and with 4-Cl, 4-OCH3 substituents at the distal phenyl ring led to high potency of the compounds. The presence of 8-Cl, 8-NO2, 8-CH3 substituents in the benzothiazole ring and H, 4-OCH3 substituents in the distal phenyl ring was responsible for marked anti-nociceptive activity. However, substitution at 8-CH3 in the benzothiazole ring with 2-Cl substitution in the distal phenyl ring resulted in a significant decrease in activity.
Acknowledgement
The financial assistance of the University Grants Commission (UGC), New Delhi, is gratefully acknowledged.
Declaration of interest: The authors declare no conflicts of interest involved in the study.
References
- Hess R. In: Frey HH, Janz D, eds. Antiepileptic Drugs. Berlin: Springer-Verlag, 1985:35.
- Brodie MJ. Lancet 1990;336:350–4.
- Chulak I, Sutorius V, Sekerka VL. Chem Pap 1990;44:131–8.
- Lacova M, Chavancova J, Hyblova O, Varkonda S. Chem Pap 1991;45:411–8.
- Papenfuhs T. Ger Offen De 1987;3:528–32.
- Huseyin U, Vanderpoorten K, Cacciaguerra S, Spampinato S, Stables JP, Depovere P, et al. J Med Chem 1998;41:1138–145.
- Jimonet P, Francois A, Barreau M, Blanchard JC, Boirean A. Eur J Med Chem 1991;42:2828–43.
- Porter RJ, Cereghiao JJ, Gladding GD, Hessie BJ, White B. Cleveland Clin Q 1984;51:293–305.
- Pandeya SN, Kohli S, Siddiqui N, Polish J. Pharmacology 2003;55:565–71.
- Siddiqui N, Pandeya SN, Khan SA, Stables JP, Rana A, Alam M, et al. Bioorg Med Chem Lett 2007;17:255–9.
- Siddiqui N, Pandeya SN, Sen AP, Singh GS. Pharmakeftiki 1992;4:121–4.
- Siddiqui N, Rana A, Khan SA, Bhat MA, Haque SE. Bioorg Med Chem Lett 2007;17:4178–82.
- Rana A, Siddiqui N, Khan SA, Haque SE, Bhat MA. Eur J Med Chem 2008;43:1114–22.
- Foye WO, Thomas LL, David AW. Principles of Medicinal Chemistry, 4th ed. Baltimore, MD: Williams & Wilkins, 1995:335.
- Sawhney SN, Bhutani S, Dharamvir. Indian J Chem 1987;26B:348–50.
- Dogruer DS, Unlu S, Sahin MF, Yesilada E. Farmaco 1998;53:80–4.
- Krall RL, Penry JK, White BG, Kupferberg HJ, Swinyard EA. Epilepsia 1978;19:409–28.
- Silvina MT, Sung CM, Luis EB, Guillermina LE. Bioorg Med Chem 2004;12:3857–69.
- Kendall DA, Browner M, Enna SJ. J Pharmacol Exp Ther 1982;220:482–7.
- Yogeeswari P, Sriram D, Saraswat V, Vaigunda RJ, Mohan KM, Murugesan S, et al. Eur J Pharm Sci 2003;20:341–6.
- Luna LG. Manual of Histological Staining Methods of the Armed Forces Institute of Pathology, 3rd ed. New York: McGraw-Hill, 1968:567.
- Lien EJ, Liuo RCH, Shinoucla HG. J Pharm Sci 1979;68:463–5.
- Leahy DE. QSAR: Rational Approaches to the Design of Bioactive Compounds. Amsterdam: Elsevier, 1991:75–82.
- Reitman S, Frankel SA. Am J Clin Pathol 1957;28:56.
- Tietz N. Fundamentals of Clinical Chemistry. St Louis, MO: WB Saunders Company, 1957:447–9.
- Toro G, Ackermann PG. Practical Clinical Chemistry, 1st ed. New York: Little, Brown and Company, 1975:210–16.
- King EJ, Armstrong AR. Can Med Assoc J 1934;31:376–81.
- Reinhold JG. In: Reiner M, ed. Standard Methods in Clinical Chemistry, 1st ed. New York: Academic Press, 1953:88–90.
- Varley H. Practical Clinical Biochemistry, 1st ed. New Delhi: CBS Publishers and Distributors, 1988:236–8.