Abstract
Enantiomers of exo- and endo-2-norbornyl-N-n-butylcarbamates were characterized as pseudo substrate inhibitors of butyrylcholinesterase. These inhibitions discriminate enantiomers of the inhibitors and therefore show stereoselectivity for the enzyme. For inhibitions by (R)-(+)- and (S)-(–)-exo-2-norbornyl-N-n-butylcarbamates, R-enantiomer is a more potent inhibitor than S-enantiomer. But, for inhibitions by (R)-(+)- and (S)-(–)-endo-2-norbornyl-N-n-butylcarbamates, S-enantiomer is a more potent inhibitor than R-enantiomer. Optically pure (R)-(+)-exo-, (S)-(–)-exo-, (R)-(+)-endo-, and (S)-(–)-endo-2-norbornyl-N-n-butylcarbamates were synthesized from condensations of optically pure (R)-(+)-exo-, (S)-(–)-exo-, (R)-(+)-endo-, and (S)-(–)-endo-2-norborneols with n-butyl isocyanate, respectively. Optically pure norborneols were obtained from kinetic resolution of their racemic esters by lipase catalysis in organic solvent.
Introduction
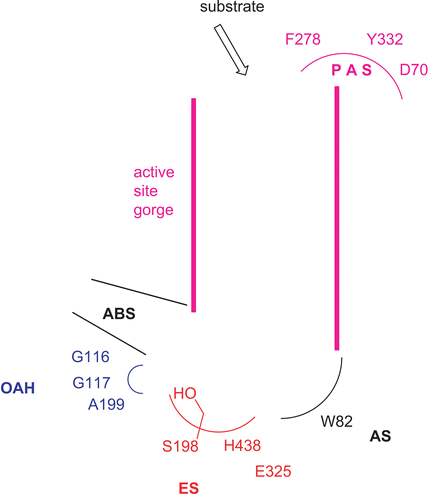
Butyrylcholinesterase (BChE, EC 3.1.1.8) is a serine hydrolase related to acetylcholinesterase (AChE, EC 3.1.1.8). Unlike AChE, which plays a vital role in the central and peripheral nervous systems, the physiological function of BChE remains unclearCitation1,2. Despite having no identified endogenous substrate, BChE plays a key role in detoxification, by degrading esters such as succinylcholine and cocaineCitation3. From X-ray crystallography, the active site structure of BChE is very similar to that of AChECitation4,5. Similar to AChECitation6–9, the active site of BChE () contains (1) an esteratic site (ES) comprising the catalytic triad Ser198-His438-Glu325, which is located at the bottom of the gorge, (2) an oxyanion hole (OAH) composed of Gly116, Gly117, and Ala199, which stabilizes the tetrahedral intermediate, (3) an anionic substrate binding site (AS) composed of Trp82, where the quaternary ammonium pole of butyrylcholine (BCh) and various active site ligands binds through a preferential interaction of quaternary nitrogens with the π electrons of aromatic groups, (4) an acyl group binding site (ABS) that binds the acyl or carbamyl group of the substrate or inhibitor, and (5) a peripheral anionic binding site (PAS)Citation10–13 composed of Phe278, Tyr332Citation14, and Asp70, which is located at the entrance (mouth) of the active site gorge and may bind to tacrine-based hetero-bivalent ligandsCitation15 and cage aminesCitation16.
Figure 1. Active sites of human BChECitation4. The enzyme active sites consist of at least five major binding sites: (1) an oxyanion hole (OAH); (2) an esteratic site (ES) or catalytic triad; (3) an anionic substrate binding site (AS); (4) an acyl binding site (ABS); and (5) a peripheral anionic binding site (PAS).
In Alzheimer’s disease (AD), a neurological disorder, cholinergic deficiency in the brain has been reportedCitation17,Citation18. Four drugs for the treatment of AD, tacrine (Cognex), donepezil (Aricept), rivastigmine (Exelon) (), and galantamine (Reminyl), are dual inhibitors of AChE and BChECitation18. The additional demonstration that central BChE rather than AChE inhibition is the best correlate of cognitive improvement in AD clinical studies with the dual cholinesterase inhibitor rivastigmine () further suggests that BChE represents an intriguing target to develop drugs for the treatment of neurodegenerative diseaseCitation19–21. The derivatives of physostigmine () are also potential drugs for the treatment of ADCitation22,Citation23. Since rivastigmineCitation24, bambuterolCitation25, and physostigmine are carbamates, inhibition mechanisms of both AChE and BChE by carbamates may play important roles for the treatment of ADCitation19–27.
Figure 2. Structures of (R)-(+)-exo-, (S)-(–)-exo-, (R)-(+)-endo-, (S)-(–)-endo-2-norbornyl-N-n-butylcarbamates, rivastigmine, physostigmine, and carbaryl.
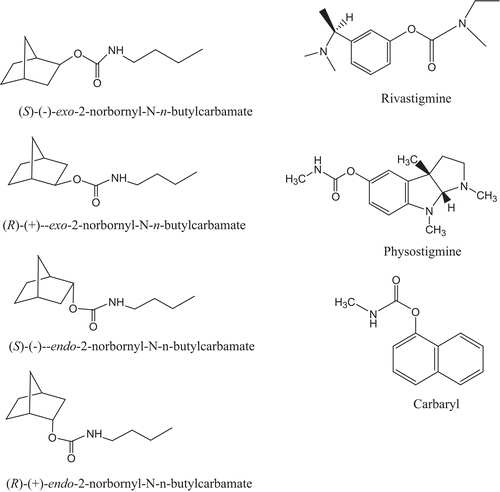
Carbaryl (1-naphthyl N-methylcarbamate, Sevin) (), carbofuran (Furadan), propoxur (Baygon), and aldicarb (Temik) are carbamate pesticides that have activities against a broad range of insects and low mammalian toxicityCitation28. These carbamate pesticides are also dual inhibitors of AChE and BChE. Therefore, inhibition mechanisms of both AChE and BChE by carbamates may also play important roles in understanding the mechanism of pesticide toxicology. Many terpenoids are reported as inhibitors of AChE and BChECitation29,30. From these reports, AChE does not show significant stereoselectivity for enantiomers of many bicyclic monoterpenoids.
The aim of this work was to study the stereoselective inhibition of BChE by chiral norbornylcarbamates. We have reported that racemic (±)-exo- and (±)-endo-2-norbornyl-N-n-butyl-carbamates are potent pseudo substrate inhibitors of BChECitation31. In this work, we further synthesized optically pure (R)-(+)-exo-, (S)-(–)-exo-, (R)-(+)-endo-, and (S)-(–)-endo-2-norbornyl-N-n-butylcarbamates from optically pure (R)-(+)-exo-, (S)-(–)-exo-, (R)-(+)-endo-, and (S)-(–)-endo-2-norborneols. These optically pure 2-norborneols were prepared from the kinetic resolution of their racemic esters by lipase in organic solventCitation32,33.
Materials and methods
Materials
Horse serum BChE (Sigma C7512), porcine pancreatic lipase (Sigma L3126), butyrylthiocholine chloride (BTCh), and 5,59-dithio-bis(-2-nitrobenzoic acid) (DTNB) were obtained from Sigma (USA). (±)-exo- and (±)-endo-2-norborneol, n-butyl isocyanate, triethylamine, CDCl3, tetramethylsilane, t-butyl methyl ether, butyryl chloride, pyridine, and (S)-(+)-α-methoxy-α-trifluoromethylphenylacetyl chloride (Mosher’s acid chloride) were purchased from Aldrich (USA). Silica gel and thin layer chromatography (TLC) plate were obtained from Merck (Germany). Hexane, CH2Cl2, ethyl acetate, and tetrahydrofuran were obtained from Tedia (USA). Sodium dihydrogen phosphate (NaH2PO4:2H2O), disodium hydrogen phosphate (Na2HPO4:12H2O), hydrogen chloride (HCl), sodium hydroxide (NaOH), potassium hydroxide (KOH), calcium chloride (CaCl2), and sodium chloride (NaCl) were purchased from UCW (Taiwan). Ethanol (95%) was obtained from Taiwan Tobacco & Liquid Corporation (Taiwan).
Instrumental methods
All steady state kinetic data were obtained from an ultraviolet (UV)-visible spectrophotometer (Agilent 8453) with a cell holder circulated by a water bath. 1H, 13C, and 19F nuclear magnetic resonance (NMR) spectra were recorded in CDCl3 at 400, 100, and 377 MHz, respectively, with internal reference tetramethylsilane (TMS) at 25°C on a Varian Gemini 400 spectrometer. Mass spectra were recorded at 71 eV in a mass spectrometer (Jeol JMS-SX/SX 102A). Elemental analyses were performed on a Heraeus instrument. Optical rotation was recorded on a polarimeter (PerkinElmer 241).
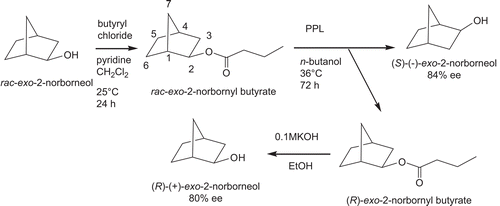
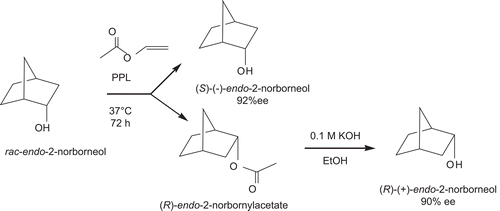
Kinetic resolution of exo- and endo-2-norborneols by lipase ( and )
(S)-(–)-exo- and (R)-(+)-exo-2-norborneol ()
To a t-butyl methyl ether (100 mL) solution of racemic (±)-exo-2-norbornyl butyrate (1 mmol) (synthesis from condensation of (±)-exo-2-norborneol with 1.2 eqs. of butyryl chloride in the presence of pyridine in CH2Cl2, 90–95% yield), porcine pancreatic lipase (4 g) was added. The reaction mixture was shaken at 36°C at 200 rpm for 72 h. This reaction yielded (S)-(–)-exo-2-norborneol (49% yield) (mp = 125–126°C and [α]D25 = –2.70°) ([α]D25 = –3.07° and mp = 126–127°C from the literatureCitation34–38) and recovered unreactive (R)-exo-2-norbornyl butyrate (51% yield). (R)-(+)-exo-2-norborneol (mp = 125–126°C and [α]D25 = +2.70°) ([α]D25 = +3.06° and mp = 126–127°C from the literatureCitation34–38) was obtained from basic hydrolysis (0.1 M KOH) of (R)-exo-norbornyl butyrate in ethanol (95%) in 99% yield.
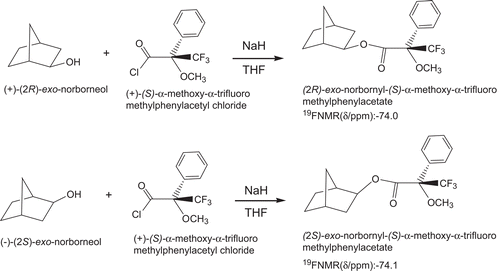
The enantiomeric excess (e.e.) values of (R)-(+)-exo- and (S)-(–)-exo-2-norborneols from the resolutions were calculated to be 80 and 84%, respectively, from the 19F-NMR spectra of their Mosher’s esters, according to the followingCitation39–41 ( and ). In an NMR tube, the condensation reaction of (R)-(+)-exo-2-norborneol (5 mM) with Mosher’s chiral derivatizing agent (S)-(+)-α-methoxy-α-trifluoromethylphenylacetyl chlorideCitation39 (5 mM) in CDCl3 in the presence of pyridine (5 mM) was carried out at 25°C for 24 h (). The fluorine chemical shifts at −73.948 and −74.113 ppm with an integration ratio of 9/1 were assigned to the fluorine atoms of (2R)- and (2S)-exo-norbornyl-(S)-α-methoxy-α-trifluoromethylphenyl acetates, respectively () (). Therefore, the enantiomeric excess of (R)-(+)-exo-2-norborneol from kinetic resolution by lipase catalysis () was calculated to be 80% from integration of these two peaks ().
Figure 3. 19F-NMR spectra after the reaction of (A) (R)-(–)-exo-2-norborneol with S-(+)-α-methoxy-α-trifluoromethylphenylacetyl chlorideCitation39 in the presence of pyridine in CDCl3 and (B) (S)-(–)-exo-2-norborneol with (S)-(+)-α-methoxy-α-trifluoromethylphenylacetyl chloride in the presence of pyridine in CDCl3. For (A), −72.069−ppm was the fluorine chemical shift of unreactive (S)-(+)-α-methoxy-α-trifluoromethylphenylacetyl chloride. The peaks at −73.948 and −74.113 ppm were assigned to the fluorine chemical shifts of (2R)- and (2S)-exo-norbornyl-(S)-α-methoxy-α-trifluoromethylphenylacetates, respectively (). For (B), −72.089 ppm was the fluorine chemical shift of unreactive S-(+)-α-methoxy-α-trifluoromethylphenylacetyl chloride. The peaks at −73.965 and −74.130 ppm were assigned to the fluorine chemical shifts of (2R)- and (2S)-exo-norbornyl-(S)-α-methoxy-α-trifluoromethylphenylacetates, respectively ().
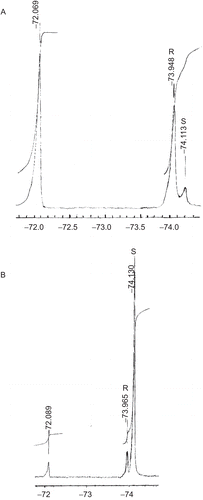
Table 1. Enantiomeric excess (%) and optical purity (%) for the kinetic resolution of racemic exo-2-norborneol () and endo-2-norborneol () by lipase in organic solvent.
(S)-(–)-exo-2-norborneol (5 mM) was also condensed with Mosher’s chiral derivatizing agent (S)-(+)-α-methoxy-α-trifluoromethylphenylacetyl chlorideCitation39 (5 mM) in CDCl3 in the presence of pyridine (5 mM) at 25°C for 24 h (). After reaction, the peaks at −73.965 and −74.130 ppm with an integration ratio of 92/8 were assigned to the fluorine atoms of (2R)- and (2S)-exo-norbornyl-(S)-α-methoxy-α-trifluoromethylphenyl acetates, respectively () (). Therefore, the enantiomeric excess of (R)-(+)-exo-2-norborneol from kinetic resolution by lipase catalysis () was calculated to be 84% ().
(S)-(–)-endo- and (R)-(+)-endo-2-norborneol ()
To a t-butyl methyl ether (50 mL) solution of racemic (±)-endo-2-norborneol (44.6 mmol) and vinyl acetate (10 mL), porcine pancreatic lipase (30 g) was added. The reaction mixture was shaken at 37°C at 200 rpm for 72 h. This reaction yielded (R)-(+)-endo-2-norbornyl acetate (49%) and recovered unreactive (S)-(–)-endo-norborneol (51%) (mp = 148–150°C and [α]D25 = –1.81°; mp = 151–152°C and [α]D25 = –1.89° from the literatureCitation34–38). (R)-(+)-endo-2-norborneol (mp = 148–150°C and [α]D25 = +1.81; [α]D25 = +1.89° and mp = 151–152°C from the literatureCitation34–38) was obtained from basic hydrolysis (0.1 M KOH) of (R)-endo-norbornyl butyrate in ethanol (95%) in 99% yield. The enantiomeric excess (e.e.) values of (S)-(–)-endo- and (R)-(+)-endo-2-norborneols from the resolutions were calculated to be 90 and 92%, respectively, from the 19F-NMR spectra of their Mosher’s esters ().
In an NMR tube, the condensation reaction of (R)-(+)-endo-2-norborneol (5 mM) with Mosher’s chiral derivatizing agent (S)-(+)-α-methoxy-α-trifluoromethylphenylacetyl chlorideCitation39 (5 mM) in CDCl3 in the presence of pyridine (5 mM) was carried out at 25°C for 24 h. The fluorine chemical shifts at −73.975 and −74.152 ppm with an integration ratio of 95/5 were assigned to the fluorine atoms of (2R)- and (2S)-endo-norbornyl-(S)-α-methoxy-α-trifluoromethylphenyl acetates, respectively. Therefore, the enantiomeric excess of (R)-(+)-endo-2-norborneol from kinetic resolution by lipase catalysis () was calculated to be 90% from integration of these two peaks ().
(S)-(–)-endo-2-norborneol (5 mM) was condensed with Mosher’s chiral derivatizing agent (S)-(+)-α-methoxy-α-trifluoromethylphenylacetyl chlorideCitation39 (5 mM) in CDCl3 in the presence of pyridine (5 mM) at 25°C for 24 h. The fluorine chemical shifts at −74.026 and −74.185 ppm with an integration ratio of 96/4 were assigned to the fluorine atoms of (2R)- and (2S)-endo-norbornyl-(S)-α-methoxy-α-trifluoromethylphenyl acetates, respectively. Therefore, the enantiomeric excess of (S)-(–)-endo-2-norborneol from kinetic resolution by lipase catalysis () was calculated to be 92% from integration of these two peaks ().
Synthesis of (R)-(+)-exo-, (S)-(–)-exo-, (R)-(+)-endo-, and (S)-(–)-endo-2-norbornyl-N-n-butylcarbamates
(R)-(+)-exo-, (S)-(–)-exo-, (R)-(+)-endo-, and (S)-(–)-endo-2-norbornyl-N-n-butylcarbamates were synthesized from condensation of optically pure (R)-(+)-exo-, (S)-(–)-exo-, (R)-(+)-endo-, and (S)-(–)-endo-2-norborneols, respectively, with 1.2 eqs. of n-butyl isocyanate in the presence of 1.2 eqs. of triethylamine in tetrahydrofuran at 25°C for 1 day (85–92% yield). All products were purified by liquid chromatography (silica gel, hexane–ethyl acetate) and were characterized by 1H- and 13C-NMR spectra, mass spectra, and elemental analysis as follows.
(R)-(+)-exo- and (S)-(–)-exo-2-norbornyl-N-n-butylcarbamates
1H-NMR (CDCl3) δ 0.92 (t, J = 7 Hz, 3H, carbamate ω-CH3), 1.40 (sextet, J = 7 Hz, 2H, carbamate γ-CH2), 1.0–1.6 (m, 7H, 4,5,6,7-norbornyl Hs), 1.56 (quintet, J = 7 Hz, 2H, carbamate β-CH2), 1.70 (m, 1H, norbornyl C(1)H), 2.24 (m, 2H, norbornyl C(3)H2), 3.15 (t, J = 7 Hz, 2H, carbamate α-CH2), 4.53 (m, 1H, norbornyl-C(2)H). 13C-NMR (CDCl3) δ 13.7 (carbamate ω-CH3), 19.9 (carbamate β-CH2), 24.2 (norbornyl C-6), 28.1 (norbornyl C-5), 32.1 (carbamate γ-CH2), 35.2 (norbornyl C-7), 35.3 (norbornyl C-4), 39.6 (norbornyl C-3), 40.6 (norbornyl C-1), 41.6 (carbamate α-CH2), 77.7 (norbornyl C-2), 156.4 (carbamate C=O). Mass spectra, exact mass: 211.157; elemental analysis: calculated for C12H21NO2: C, 68.21; H, 10.02; N, 6.63, found C, 68.15; H, 10.32; N, 6.56%. mp 178–180°C) (decomp.).
(R)-(+)-endo- and (S)-(–)-endo-2-norbornyl-N-n-butylcarbamates
1H-NMR (CDCl3) δ 0.92 (t, J = 7 Hz, 3H, carbamate ω-CH3), 1.20–1.80 (m, 11H, carbamate β- and γ-CH2 and 4,5,6,7-norbornyl Hs), 1.96 (m, 1H, norbornyl C(1)H), 2.10–2.50 (m, 2H, norbornyl C(3)H2), 3.19 (t, J = 7 Hz, 2H, carbamate α-CH2), 4.60 (br. s, 1H, carbamate NH), 4.89 (m, 1H, norbornyl-C(2)H). 13C-NMR (CDCl3) δ 13.7 (carbamate ω-CH3), 19.8 (carbamate β-CH2), 20.9 (norbornyl C-6), 29.4 (norbornyl C-5), 32.1 (carbamate γ-CH2), 36.4 (norbornyl C-7), 36.9 (norbornyl C-4), 37.2 (norbornyl C-3), 40.4 (carbamate α-CH2), 40.7 (norbornyl C-1), 75.7 (norbornyl C-2), 156.8 (carbamate C=O). Mass spectra, exact mass: 211.157; elemental analysis: calculated for C12H21NO2: C, 68.21; H, 10.02; N, 6.63, found C, 68.17; H, 10.30; N, 6.58%. mp 178–180°C (decomp.).
Data reduction
Origin (version 6.0) was used for linear and nonlinear least-squares curve fittings.
Enzyme inhibition
BChE inhibitions by the carbamate inhibitors were assayed by the Ellman methodCitation42. BChE-catalyzed hydrolysis of BTCh in the presence of the carbamate inhibitors and DTNB was followed continuously at 410 nm on a UV-visible spectrometer. The temperature was maintained at 25.0°C by a refrigerated circulating water bath. All inhibition reactions were performed in sodium phosphate buffer (1 mL, 0.1 M, pH 7.0) containing NaCl (0.1 M), acetonitrile (2% by volume), triton X-100 (0.5% by weight), substrate (ATCh for AChE or BTCh for BChE) (50 μM), DTNB, and varying concentrations of inhibitor. Requisite volumes of stock solution of substrate and inhibitors in acetonitrile were injected into the reaction buffer via a pipet. BChE was dissolved in sodium phosphate buffer (0.1 M, pH 7.0). The first-order rate constant (kapp) for inhibition was determined as described by Hosie et al.Citation43–47. The apparent inhibition constant ((1 + [S]/Km)Ki) and carbamylation constant (k2) were obtained from the nonlinear least-squares curve fitting of the kapp vs. [I] plot following Equation (1) (). Duplicate sets of data were collected for each inhibitor concentration.
Figure 4. Nonlinear least-squares curve fittings of (A) the kapp vs. (R)-(+)-exo-2-norbornyl-N-n-butylcarbamate concentration ([I]) plot and (B) the kapp vs. (R)-(+)-exo-2-norbornyl-N-n-butylcarbamate concentration ([I]) plot following Equation (1) for pseudo substrate inhibition of BChE. For , the parameters of the fit were k2 = 0.0030 ± 0.0001 s−1 and (1 + Km/[S])Ki = 22 ± 4 nM with R= 0.9963. After calculation, Ki = 11 ± 2 nM and ki = (270 ± 50) × 103 M− 1 s−1 (). For , the parameters of the fit were k2 = 0.00310 ± 0.00007 s−1 and (1 + Km/[S])Ki = 100 ± 4 nM with R= 0.9920. After calculation, Ki = 50 ± 2 nM and ki = (62 ± 3) × 103 M− 1 s−1 ().
![Figure 4. Nonlinear least-squares curve fittings of (A) the kapp vs. (R)-(+)-exo-2-norbornyl-N-n-butylcarbamate concentration ([I]) plot and (B) the kapp vs. (R)-(+)-exo-2-norbornyl-N-n-butylcarbamate concentration ([I]) plot following Equation (1) for pseudo substrate inhibition of BChE. For Figure 4A, the parameters of the fit were k2 = 0.0030 ± 0.0001 s−1 and (1 + Km/[S])Ki = 22 ± 4 nM with R= 0.9963. After calculation, Ki = 11 ± 2 nM and ki = (270 ± 50) × 103 M− 1 s−1 (Table 2). For Figure 4B, the parameters of the fit were k2 = 0.00310 ± 0.00007 s−1 and (1 + Km/[S])Ki = 100 ± 4 nM with R= 0.9920. After calculation, Ki = 50 ± 2 nM and ki = (62 ± 3) × 103 M− 1 s−1 (Table 2).](/cms/asset/5c7b4660-832b-42bc-90a0-e4bebe3ecd7d/ienz_a_388992_f0004_b.gif)
Results and discussion
Kinetic resolution of norborneols by lipase catalysis
In this article, we first report the kinetic resolution of (R)-(+)-exo-, (S)-(–)-exo-, (R)-(+)-endo-, and (S)-(–)-endo-2-norborneols by lipase catalysis in organic solvent ( and ). The absolute configurations of (S)-(–)-exo-, (R)-(+)-exo-, (S)-(–)-endo-, and (R)-(+)-endo-2-norborneols were determined on the basis of their optical rotation valuesCitation34–38 and the 19F-NMR spectra of their Mosher’s ester derivativesCitation39–41 ( and ).
Scheme 1. Kinetic resolution of (R)-(+)- and (S)-(–)-exo-2-norborneols from lipase-catalyzed hydrolysis of racemic (±)-exo-2-norbornyl butyrate.
Scheme 2. Kinetic resolution of (R)-(+)- and (S)-(–)-endo-2-norborneols from lipase-catalyzed acetylation of racemic (±)-exo-2-norborneol with vinyl acetate.
Scheme 3. Determination of enantiomeric excess and absolute configuration of (R)-(+)- and (S)-(–)-exo-2-norborneols by 19F-NMR spectra of their Mosher’s ester derivatives.
Pseudo substrate inhibitions of BChE by carbamates
The mechanism for BChE-catalyzed hydrolysis of substrate is formation of the first tetrahedral intermediate via nucleophilic attack of the active site Ser () to substrate, then formation of the acyl enzyme intermediate from the intermediate (). In the presence of substrate, (R)-(+)-exo-, (S)-(–)-exo-, (R)-(+)-endo-, and (S)-(–)-endo-2-norbornyl-N-n-butylcarbamates are characterized as the pseudo or alternateCitation48,49 substrate inhibitors of BChE, like many other carbamates (, , and )Citation43–47. Presumably, the carbonyl carbons of the n-butylcarbamyl moieties of inhibitors are nucleophilically attacked by the active site serine of the enzyme then formation of the n-butylcarbamyl enzyme follows ().
Scheme 4. Kinetic scheme for pseudo substrate inhibition of BChE by 2-norbornyl-N-n-butylcarbamate in the presence of substrate. E, enzyme; E-A, acyl enzyme; EI, enzyme–inhibitor Michaelis complex; E-I9, carbamyl enzyme; ES, enzyme–substrate Michaelis complex; I, pseudo substrate inhibitor; k2, carbamylation constant; k3, decarbamylation constant; k2S, formation rate constant of E-A; k3S, deacylation constant of E-A; Ki, inhibition constant; Km, Michaelis–Menten constant; P, product, 2-norborneol; P9, product, thiocholine; P99, product, butyrate; Q, product, butylcarbamic acid; S, substrate, BTCh.
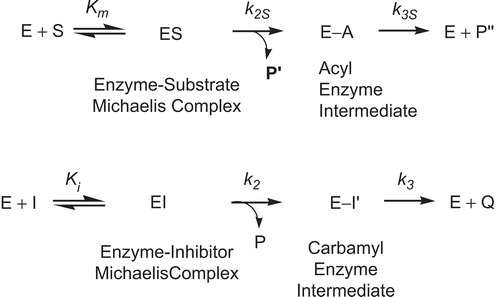
Table 2. The k2, Ki, and ki valuesa of BChE inhibitions by stereoisomers of 2-norbornyl carbamates.
BChE inhibitions by (R)-(+)- and (S)-(–)-exo-2-norbornyl-N-n-butylcarbamates
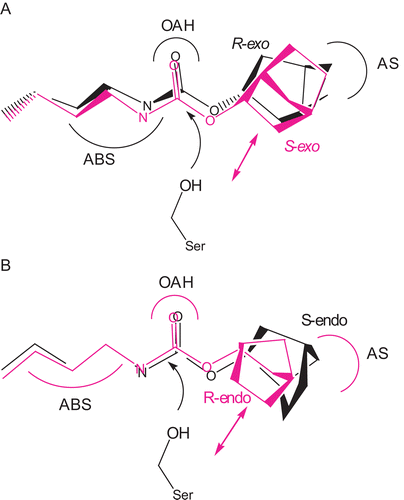
Comparing the BChE inhibitory potency between both enantiomers of exo-2-norbornyl-N-n-butylcarbamate, R-enantiomer is about 3.35 times more potent than S-enantiomer (). Therefore, BChE shows stereoselectivity for (R)-(+)-exo-2-norbornyl-N-n-butylcarbamate over (S)-(–)-exo-2-norbornyl-N-n-butylcarbamate. Ki and ki values for the racemic inhibitor are about the average of those of both enantiomers (). This stereopreference (R < S) is the same as those reported for the BChE-catalyzed hydrolysis enantiomers of quinuclidin-3-acetateCitation50, bambuterolCitation51, quinuclidinium benzoateCitation52, and cocaineCitation53. Modeling both enantiomers of exo-2-norbornyl-N-n-butylcarbamate into the active site of BChECitation4,5 () indicates that the norbornyl ring of (R)-enantiomer fits better into the AS of the enzyme than that of (S)-enantiomer (). On the other hand, an unfavorable repulsive force between the norbornyl ring of (S)-enantiomer and the nucleophilic serine of the enzyme has been observed in this model (). Therefore, BChE stereoselectively hydrolyzes R-enantiomers of the exo-substituted ring compounds such as exo-2-norbornyl-N-n-butylcarbamate, quinuclidin- 3-acetateCitation50, quinuclidinium benzoateCitation52, and cocaineCitation53.
Figure 5. Superimposition of (A) (R)-(+)- and (S)-(–)-exo-2-norbornyl-N-n-butyl- carbamates and (B) (R)-(+)- and (S)-(–)-endo-2-norbornyl-N-n-butylcarbamates at their carbamyl moieties and fitting both enantiomers into the active site of BChE. For (A), an unfavorable repulsion between S-enantiomer and the active serine of the enzyme was observed. For (B), an unfavorable repulsion between R-enantiomer and the active serine of the enzyme was observed.
BChE inhibitions by (R)-(+)- and (S)-(–)-endo-2-norbornyl-N-n-butylcarbamates
Comparing the BChE inhibitory potency between both enantiomers of endo-2-norbornyl-N-n-butylcarbamate, S-enantiomer is 2.5 times more potent than R-enantiomer (). Therefore, BChE shows setereoselectivity for (S)-(–)-endo-2-norbornyl-N-n-butylcarbamate over (R)-(+)-endo-2-norbornyl-N-n-butylcarbamate. Ki and ki values for the racemic inhibitor are about the average of those of both enantiomers (). However, this stereopreference (S < R) is opposite to those reported for the BChE-catalyzed hydrolysis enantiomers of quinuclidin-3-acetateCitation50, bambuterolCitation51, quinuclidinium benzoateCitation52, and cocaineCitation53. Modeling both enantiomers of endo-2-norbornyl-N-n-butylcarbamate into the active site of the X-ray structure of BChECitation4,Citation5 () suggests that the norbornyl ring of (S)-enantiomer fits better into the AS of the enzyme than (R)-enantiomer (). On the other hand, an unfavorable repulsive force between the norbornyl ring of (R)-enantiomer and the nucleophilic serine of the enzyme is proposed in this model (). The different stereopreference of BChE for endo-2- norbornyl-N-n-butylcarbamate may be due to the fact that the AS site of BChE strongly binds to C-7 of endo-2-norbornyl-N-n-butylcarbamate instead ().
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Massoulie J, Pezzementi L, Bon S, Krejci E, Vallette FM. Prog Neurobiol 1993;41:31–91.
- Xie W-H, Stribley JA, Chatonnet A, Wilder PJ, Rizzino A, McComb RD, et al. Pharmacol Exp Ther 2000;293:869–902.
- Gorelick DA. Drug Alcohol Depend. 1997; 48:159–165.
- Nicolet Y, Lockridge O, Masson P, Fontecilla-Camps JC, Nachon F. J Biol Chem 2003; 278:41141–7.
- Loudwig S, Nicolet Y, Masson P, Fontecilla-Camps JC, Bon S, Nachon F, et al. ChemBioChem 2003; 4:762–7.
- Sussman JL, Harel M, Frolow F, Oefner C, Goldma A, Toker L, et al. Science 1991; 253:872–9.
- Harel M, Sussman JL, Krejci E, Bon S, Chanal P, Massoulie J, et al. Proc Natl Acad Sci USA 1992; 89:10827–31.
- Bar-On P, Millard CB, Harel M, Dvir H, Enz A, Sussman JL, et al. Biochemistry 2002; 41:3555–63.
- Harel M, Quinn DM, Nair HK, Silman I, Sussman JL. J Am Chem Soc 1996; 118:2340–6.
- Radic Z, Pickering NA, Vellom DC, Camp S, Taylor P. Biochemistry 1993; 32:12074–84.
- Taylor P, Mayer RT, Himel CM. Mol Pharmacol 1994;45:74–83.
- Pang Y-P, Quiram P, Jelacic T, Hong F, Brimjoin S. J Biol Chem 1996;271:23646–9.
- Lin G, Lai C-Y, Liao W-C. Bioorg Med Chem 1999;7:2683–9.
- Savini L, Gaeta A, Fattorusso C, Catalanotti B, Campiani G, Chiasserini L, et al. J Med Chem 2003;46:1–4.
- Masson P, Xie W, Forment M-T, Levitsky V, Fortier P-L, Albaret C, et al. Biochim Biophys Acta 1999; 1433:281–93.
- Lin G, Tsai H-J, Tsai Y-H. Bioorg Med Chem Lett 2003;13:2887–90.
- Mesulam M. In: Giacobini E, ed. Cholinesterase and Cholinesterase Inhibitors. London: Martin Dunitz, 2000. 121–137.
- Giacobini E. Int J Geriatr Psychiatry 2003;18:S1–5.
- Greig NH, Utsuki T, Yu Q-S, Zhu X, Holloway HW, Perry T, et al. Curr Med Res Opin 2001; 17:1–6.
- Mesulam MM, Guillozet A, Shaw P, Levey A, Duysen EG, Lockridge O. Neuroscience 2002; 110:627–39.
- Giacobini E, Spiegel R, Enz A, Veroff AE, Cutler NR. J Neural Transm 2002; 109:1053–65.
- Yu Q-S, Holloway HW, Flippen-Anderson JL, Hoffman B, Brossi A, Greig NH. J Med Chem 2001; 44:4062–71.
- Stojan J, Pavlic MR. J Enzyme Inhib 1997;11:199–208.
- Bartolucci C, Perola E, Cellai L, Brufani M, Lamba D. Biochemistry 1999;38:5714–19.
- Kovarik Z, Simeon-Rudolf V. J Enzyme Inhib Med Chem 2004;19:113–17.
- Lin G, Liu Y-C, Lin Y-F, Wu Y-G. J Enzyme Inhib Med Chem 2004;19:395–401.
- Lin G, Lee Y-R, Liu Y-C, Wu Y-G. Chem Res Toxicol 2005;18:1124–31.
- Baron RL. Carbamate insecticides. In: Hayes WJJ, Laws ERJ, eds. Handbook of Pesticide Toxicology. New York: Academic Press, 1991. 1125–1189.
- Nisar M, Ahmad M, Wadood N, Lodhi MA, Shaheen F, Choudhary MI. J Enzyme Inhib Med Chem 2009; 24:47–51.
- Miyazawa M, Yamafuji C. J Agric Food Chem 2005; 53:1765–8.
- Lin G, Chen G-H, Ho H-C. Bioorg Med Chem Lett 1998; 8:2747–50.
- Boland W, Frößl C, Lorenz N. Synthesis 1991; 12:1049–72.
- Theil F. Chem Rev 1995; 95:2203–27.
- Irwin AJ, Jones JB. J Am Chem Soc 1976; 98:8476–82.
- Berson JA, Walla JS, Remanick A, Suzuki S, Reynods-Warnhoff P, Willner D. J Am Chem Soc 1961; 83:3986–97.
- Nakazaki M, Chikamatsu H, Naemura K, Asao M. J Org Chem 1980; 45:4432–40.
- Winstein S, Trifan D. J Am Chem Soc 1951; 74:1154–60.
- Yoshizako F, Nishimura A, Chubachi M, Kirihata M. J Ferm Bioeng 1996; 82:601–3.
- Dale JA, Mosher HS. J Am Chem Soc 1973; 95:512–19.
- Takahashi T, Fukuishima A, Tanaka Y, Takeuchi Y, Kabuto K, Kabuto C. Chem Commun 2000:788–789.
- Takahashi T, Kameda H, Kamei T, Ishizaki M. J Fluorine Chem 2006; 127:760–8.
- Ellman CL, Courtney KD, Andres VJ, Featherstone RM. Biochem Pharm 1961; 7:88–95.
- Hosie L, Sutton LD, Quinn DM. J Biol Chem 1987; 262:260–4.
- Feaster SR, Lee K, Baker N, Hui DY, Quinn DM. Biochemistry 1996; 35:16723–34.
- Feaster SR, Quinn DM. Methods Enzymol 1997; 286:231–52.
- Lin G, Lai C-Y. Tetrahedron Lett 1995; 36:6117–20.
- Lin G, Shieh C-T, Ho H-C, Chouhwang J-Y, Lin W-Y, Lu C-P. Biochemistry 1999; 38:9971–81.
- Pietsch M, Gütschow M. J Biol Chem 2002; 277:24006–13.
- Pietsch M, Gütschow M. J Med Chem 2005; 48:8270–88.
- Simeon-Rudolf V, Tomic S, Bosak A, Primozic I, Orsulic M. Chem Biol Interact 2005; 157:420–1.
- Bosak A, Gazic I, Vinkovic V, Kovarik Z. Chem Biol Intact 2008; 175:192–5.
- Primozic I, Hrenar T, Tomic S, Meic Z. European J Org Chem 2003;(2):295–301.
- Zhan C-G, Zheng F, Landry DW. J Am Chem Soc 2003; 125:2462–74.