Abstract
In this study, some 4-(1,5-diarylimidazol-2-yl)thioacetyl-1-phenyl-2,3-dimethyl-3-pyrazoline-5-one derivatives were prepared by reacting 4-(2-chloroacetyl)-1-phenyl-2,3-dimethyl-3-pyrazoline-5-one and 2-mercapto-1, 5-diarylimidazole derivatives. The antinociceptive and anticancer activities of the compounds obtained were investigated. It was observed that some of the compounds, 2a, 2d, 2g, and 2j, showed remarkable antinociceptive activity, and one of the compounds, 2i, showed weak anticancer activity.
Introduction
The antinociceptive and antiinflammatory activities of 1,2-diaryl heterocyclics by cyclooxygenase-2 (COX-2) inhibition have been known since the 1970sCitation1–4. Since then, extensive research in this area has been carried out, and numerous compounds have been synthesized in a search for COX-2 inhibitory, antinociceptive and antiinflammatory activities. Two of these compounds, namely celecoxib I and rofecoxib II (), have been employed clinically. However, these drugs have been found to have serious side effects on the heart.
Figure 1. Structures of celecoxib I and rofecoxib II.
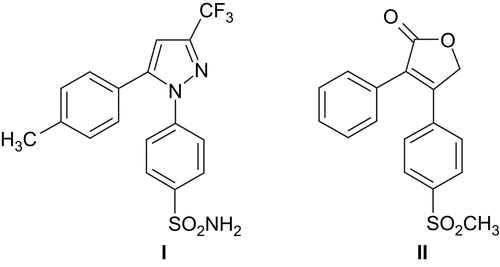
The heterocyclic residue of these compounds may be five- or six-membered, such as furan, thiophene, pyrrole, oxazole, thiazole, imidazole, pyrazole, pyridine, pyrimidine, etc.Citation5–20. The pyrazolones, especially antipyrine (1-aryl-2, 3-dimethyl-3-pyrazoline-4-one) derivatives, are well known for their antinociceptive and antipyretic activities and have been used widely in the clinicCitation21. In light of the above findings, it appears that both 1,2-diarylheterocyclic and antipyrine residues are two important active pharmacophoric structures for antinociceptive activity. Besides, it has been well documented that 1,2-diaryl heterocyclic compounds have cytotoxic activity as a result of COX-2 inhibitionCitation1,Citation22–26.
In the present study, we have aimed to incorporate 1, 5-diarylimidazole and antipyrine residues in a single molecule and investigate antinociceptive and anticancer activities of the resultant compounds.
Experimental
Chemistry
Melting points were determined by using an Electrothermal 9100 digital melting point apparatus and were uncorrected. Spectroscopic data were recorded on the following instruments: FTIR (Fourier transform infrared), Shimadzu 8400S spectrophotometer; 1H-NMR (nuclear magnetic resonance), Bruker 500 NMR spectrometer. Analyses for C, H, and N were within 0.4% of the theoretical values.
4-(2-Chloroacetyl-1-phenyl-2,3-dimethyl-3-pyrazoline-5-oneCitation27 and 2-mercapto-1,5-diarylimidazolesCitation28 were prepared according to the literature methods. Some characteristics of the compounds are given in .
Table 1. Some characteristics of the compounds.
General methods for preparation of 4-(1, 5-diarylimidazol-2-yl)thioacetyl-1-phenyl-2, 3-dimethyl-3-pyrazoline-5-one derivatives
A mixture of 4-(2-chloroacetyl-1-phenyl-2,3-dimethyl-3-pyrazoline-5-one (5 mmol, 1.32 g), the appropriate 2- mercapto-1,5-diarylimidazole derivative (5.5 mmol), and K2CO3 (6 mmol, 0.83 g) in acetone was refluxed for 8 h. The excess acetone was evaporated. The residue was washed with water and recrystallized from ethanol.
2a IR (KBr) νmax (cm−1): 1639 (C = O), 1593−1506 (C = C), 1H-NMR (500 MHz) (DMSO-d6) (ppm): 2.58 (3H, s, CH3), 3.35 (3H, s, CH3), 4.48 (2H, s, CH2), 7.06 (2H, d, J: 7.84 Hz, Ar-H), 7.18–7.27 (3H, m, Ar-H), 7.28–7.30 (3H, m, Ar-H), 7.36 (2H, d, J: 7.86 Hz, Ar-H), 7.48–7.51 (4H, m, Ar-H), 7.54–7.58 (2H, m, Ar-H).
2b IR (KBr) νmax (cm−1): 1651, 1637 (C = O), 1590−1506 (C = C), 1H-NMR (500 MHz) (DMSO-d6) (ppm): 2.49 (3H, s, CH3), 2.58 (3H, s, CH3), 3.34 (3H, s, CH3), 4.49 (2H, s, CH2), 7.08 (2H, d, J: 8.01 Hz, Ar-H), 7.13–7.25 (3H, m, Ar-H), 7.27–7.30 (2H, m, Ar-H), 7.36 (2H, d, J: 8.57 Hz, Ar-H), 7.47–7.53 (4H, m, Ar-H), 7.55–7.58 (2H, m, Ar-H).
2c IR (KBr) νmax (cm−1): 1647 (C = O), 1621−1520 (C = C), 1H-NMR (500 MHz) (DMSO-d6) (ppm): 2.62 (3H, s, CH3), 3.34 (3H, s, CH3), 3.69 (3H, s, OCH3), 4.45 (2H, s, CH2), 6.80 (2H, d, J: 7.71 Hz, Ar-H), 6.99 (2H, d, J: 7.65 Hz, Ar-H), 7.17 (1H, s, Ar-H), 7.26–7.28 (2H, m, Ar-H), 7.36 (2H, d, J: 7.97 Hz, Ar-H), 7.40–7.50 (4H, m, Ar-H), 7.55–7.58 (2H, m, Ar-H).
2d IR (KBr) νmax (cm−1): 1655, 1638 (C = O), 1593−1498 (C = C), 1H-NMR (500 MHz) (DMSO-d6) (ppm): 2.58 (3H, s, CH3), 3.36 (3H, s, CH3), 4.48 (2H, s, CH2), 7.09–7.10 (4H, m, Ar-H), 7.26 (1H, s, Ar-H), 7.28–7.30 (2H, m, Ar-H), 7.36 (2H, d, J: 7.87 Hz, Ar-H), 7.48–7.52 (4H, m, Ar-H), 7.55–7.58 (2H, m, Ar-H).
2e IR (KBr) νmax (cm−1): 1635 (C = O), 1589−1514 (C = C), 1H-NMR (500 MHz) (DMSO-d6) (ppm): 2.36 (3H, s, CH3), 2.58 (3H, s, CH3), 3.34 (3H, s, CH3), 4.47 (2H, s, CH2), 7.08 (2H, d, J: 7.82 Hz, Ar-H), 7.16 (2H, d, J: 8.06 Hz, Ar-H), 7.19–7.26 (4H, m, Ar-H), 7.29 (2H, d, J: 8.00 Hz, Ar-H), 7.36 (2H, d, J: 7.96 Hz, Ar-H), 7.45–7.50 (1H, m, Ar-H), 7.55–7.58 (2H, m, Ar-H).
2f IR (KBr) νmax (cm−1): 1642 (C = O), 1605−1505 (C = C), 1H-NMR (500 MHz) (DMSO-d6) (ppm): 2.37 (3H, s, CH3), 2.49 (3H, s, CH3), 2.58 (3H, s, CH3), 3.34 (3H, s, CH3), 4.47 (2H, s, CH2), 7.08 (2H, d, J: 8.32 Hz, Ar-H), 7.16 (2H, d, J: 8.72 Hz, Ar-H), 7.18–7.23 (2H, m, Ar-H), 7.26 (1H, s, Ar-H), 7.29 (2H, d, J: 8.13 Hz, Ar-H), 7.36 (2H, d, J: 7.78 Hz, Ar-H), 7.48–7.50 (1H, m, Ar-H), 7.54–7.58 (2H, m, Ar-H).
2g IR (KBr) νmax (cm−1): 1638 (C = O), 1611−1508 (C = C), 1340, 1H-NMR (500 MHz) (DMSO-d6) (ppm): 2.37 (3H, s, CH3), 2.58 (3H, s, CH3), 3.34 (3H, s, CH3), 3. 69 (3H, s, OCH3), 4.44 (2H, s, CH2), 6.81 (2H, d, J: 8.02 Hz, Ar-H), 7.09 (2H, d, J: 8.80 Hz, Ar-H), 7.15 (2H, d, J: 7.92 Hz, Ar-H), 7.24 (1H, s, Ar-H), 7.28 (2H, d, J: 8.26 Hz, Ar-H), 7.36 (2H, d, J: 8.17 Hz, Ar-H), 7.47–7.51 (3H, m, Ar-H), 7.55–7.58 (2H, m, Ar-H).
2h IR (KBr) νmax (cm−1): 1646, 1631 (C = O), 1596−1498 (C = C), 1340, 1H-NMR(500 MHz) (DMSO-d6) (ppm): 2.36 (3H, s, CH3), 2.58 (3H, s, CH3), 3.34 (3H, s, CH3), 4.47 (2H, s, CH2), 7.10–7.12 (4H, m, Ar-H), 7.16 (2H, d, J: 8.15 Hz, Ar-H), 7.24 (1H, s, Ar-H), 7.29 (2H, d, J: 8.20 Hz, Ar-H), 7.36 (2H, d, J: 7.42 Hz, Ar-H), 7.47–7.50 (1H, m, Ar-H), 7.55–7.58 (2H, m, Ar-H).
2i IR (KBr) νmax (cm−1): 1636 (C = O), 1589−1514 (C = C), 1H-NMR (500 MHz) (DMSO-d6) (ppm): 2.58 (3H, s, CH3), 3.35 (3H, s, CH3), 3.80 (3H, s, OCH3), 4.47 (2H, s, CH2), 7.02 (2H, d, J: 8.67 Hz, Ar-H), 7.09 (2H, d, J: 7.85 Hz, Ar-H), 7.18–7.24 (5H, m, Ar-H), 7.26 (1H, s, Ar-H), 7.36 (2H, d, J: 8.07 Hz, Ar-H), 7.47–7.50 (1H, m, Ar-H), 7.55–7.58 (2H, m, Ar-H).
2j IR (KBr) νmax (cm−1): 1638 (C = O), 1595−1502 (C = C), 1340, 1H-NMR (500 MHz) (DMSO-d6) (ppm): 2.49 (3H, s, CH3), 2.58 (3H, s, CH3), 3.34 (3H, s, CH3), 3.80 (3H, s, OCH3), 4.47 (2H, s, CH2), 7.03 (2H, d, J: 8.84 Hz, Ar-H), 7.09 (2H, d, J: 8.33 Hz, Ar-H), 7.21 (2H, d, J: 8.79 Hz, Ar-H), 7.25 (1H, s, Ar-H), 7.36 (2H, d, J: 7.37 Hz, Ar-H), 7.47–7.50 (1H, m, Ar-H), 7.55–7.58 (2H, m, Ar-H).
2k IR (KBr) νmax (cm−1): 1658, 1641 (C = O), 1598−1512 (C = C), 1H-NMR (500 MHz) (DMSO-d6) (ppm): 2.58 (3H, s, CH3), 3.35 (3H, s, CH3), 3.69 (3H, s, OCH3), 3.80 (3H, s, OCH3), 4.46 (2H, s, CH2), 6.70–6.87 (4H, m, Ar-H), 7.03 (2H, d, J: 8.80 Hz, Ar-H), 7.15 (1H, s, Ar-H), 7.19 (2H, d, J: 8.77 Hz, Ar-H), 7.35 (2H, d, J: 7.62 Hz, Ar-H), 7.48–7.51 (1H, m, Ar-H), 7.55–7.58 (2H, m, Ar-H).
2l IR (KBr) νmax (cm−1): 1632 (C = O), 1589−1498 (C = C), 1H-NMR (500 MHz) (DMSO-d6) (ppm): 2.58 (3H, s, CH3), 3.35 (3H, s, CH3), 3.80 (3H, s, OCH3), 4.46 (2H, s, CH2), 7.02 (2H, d, J: 8.86 Hz, Ar-H), 7.10–7.12 (4H, m, Ar-H), 7.15 (1H, s, Ar-H), 7.21 (2H, d, J: 8.82 Hz, Ar-H), 7.23 (1H, s, Ar-H), 7.35 (2H, d, J: 8.54 Hz, Ar-H), 7.48–7.51 (1H, m, Ar-H), 7.55–7.58 (2H, m, Ar-H).
Pharmacology
Animals
All the animals were housed in cages with free access to food and water. They were placed in a quiet and temperature–humidity controlled room (22 ± 2°C and 60 ± 5%, respectively) in which a 12:12 light–dark cycle was maintained. Mice (25–30 g) of either sex were used in the experiments. The animals were divided into 13 groups. Seven or eight animals were used in each study group. The mice were allowed 1–2 h to adjust to the laboratory conditions. All compounds were given intraperitoneally (i.p.) at 100 mg/kg doses. The control animals received 0.1 mL dimethylsulfoxide (DMSO) i.p. Morphine sulfate (5 mg/kg) and dipyrone (100 mg/kg) were used as the reference antinociceptive agents. The study was approved by the Local Ethics Committee of Osmangazi University, Medical School, Eskisehir, Turkey.
Tail clip test
A pressure-standardized artery clip was applied 3–4 cm from the tip of the tail for evaluation of the response to noxious pressure. Turning toward or biting at the clip within 15 s of artery clip placement was the threshold used in this testCitation29.
Hot-plate test
The test was based on that described by Eddy and Leimbach. A transparent glass cylinder (16 cm high, 16 cm diameter) was used to keep the mouse on the heated surface of the plate. The temperature of the hot-plate was set to 55 ± 0.5°C by using a thermoregulated water-circulating pump. The time of latency was defined as the time period between the zero point when the animal was placed on the hot-plate surface and the time when the animal licked its paw or jumped off to avoid thermal pain (cutoff time 30 s)Citation30–32.
Abdominal constriction test
This test was performed by the i.p. injection of 0.6% acetic acid (60 mg/kg). The number of stretching movements (arching of back, development of tension in the abdominal muscles, elongation of the body, and extension of the forelimbs) was observed. Stretching movements commenced 5 min after acetic acid injection. These contractions were counted and recorded for 10 min. Antinociceptive activity was expressed as the reduction in the number of abdominal constrictionsCitation33.
Anticancer activity test
The cytotoxic and/or growth inhibitory effects of the compounds were evaluated in vitro against approximately 66 human tumor cell lines derived from nine neoplastic diseases, namely: leukemia (L), non-small cell lung cancer (NSCLC), colon cancer (CC), central nervous system cancer (CNSC), melanoma (M), ovarian cancer (OC), renal cancer (RC), prostate cancer (PC), and breast cancer (BC). The evaluation of anticancer activity was performed at the National Cancer Institute (NCI) of Bethesda, USA, following the in vitro screening program, which is based upon the use of multiple panels of 66 human tumor cell lines against which our compounds were tested at 10-fold dilutions of five concentrations ranging from 10−4 to 10−8 M. The percentage growth was evaluated spectrophotometrically versus controls not treated with test agents. A 48 h continuous drug exposure protocol was followed and a sulforhodamine B (SRB) protein assay was used to estimate cell viability of growthCitation34.
Results and discussion
Chemistry
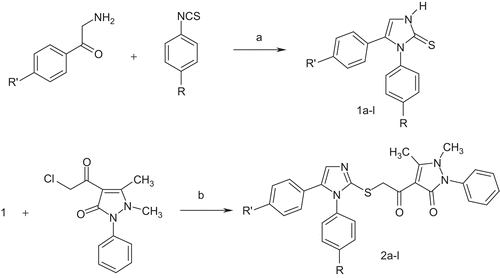
The syntheses of the title 4-(1,5-diarylimidazol-2-yl)thioacetyl-1-phenyl-2,3-dimethyl-3-pyrazoline-5-one derivatives 2a–l were accomplished in accordance with the sequence of reaction depicted in . The starting materials, 1,5-diaryl-2-mercaptoimidazoles 1a–l, were prepared by reacting the appropriate 2-amino-49-substituted acetophenones and 4-substituted phenylisothiocyanates in pyridine according to the method described in the literatureCitation28. To obtain the final products 2a–l, 4-(2-chloroacetyl-1-phenyl-2,3-dimethyl-3-pyrazoline-5-one was reacted with suitable imidazole derivatives 1a–l under Williamson ether synthesis conditions. The structures of the obtained compounds were elucidated using spectral data. In the IR spectra, the charactesistic amide and ketone carbonyl functions were observed in the 1672– 1652 cm−1 region separately or as a single bandCitation35. The NMR spectra of the compounds 2a–l exhibited singlets resulting from resonances of the thioacetyl-1-phenyl-2,3-dimethyl-3-pyrazoline-5-one residue assigned to C-CH3 protons at 2.58 ppm, to N-CH3 protons at 3.34–3.36 ppm, and to S-CH2-CO protons at 4.45–4.49 ppm, respectively. The other common proton groups existing in all compounds, imidazole-C4-H protons, were obtained as singlets at 7.26–7.29 ppm for some of the compounds. For the others, the mentioned protons were taking part in multiplets because of overlaping with aromatic protons.
Scheme 1. Synthesis of compounds 1a–l and 2a–l. Reagents and conditions: (a) pyridine, heating at reflux; (b) K2CO3, acetone.
Antinociceptive activity
Antinociceptive activities of the compounds were determined by using the tail clip test, hot-plate test, and abdominal constriction test. Both the tail clip and hot-plate tests were used to evaluate central antinociceptive activity and the abdominal constriction test was used to assess peripheral antinociceptive activity. The findings are shown in and all data were compared to control groups. The results are given as a percentage of the maximal possible effect (%MPE ± SEM), which is defined by following equation:
Table 2. Antinociceptive activity of the compounds.
Statistical analyses were carried out using Student’s t-test.
Compounds 2a, 2d, and 2g exhibited antinociceptive activity in the tail clip test. 2a and 2d showed greater antinociceptive activities than the reference compounds. The other compounds did not display any significant antinociceptive activity in this test.
In the hot-plate test, only compound 2d evoked antinociceptive activity when compared with the control group or dipyrone. On the other hand, the other compounds did not exhibit any antinociceptive activity when compared with control or reference compounds.
In the abdominal constriction test, although all the compounds exhibited significant antinociceptive activities, compounds 2d and 2j especially were found to produce the most antinociceptive activity at the dose tested. Compound 2d was antinociceptive when compared with dipyrone (p ≤ 0.05), and at 100 mg/kg it produced antinociceptive activity equivalent to that of morphine. 2a was the only compound not to show antinociceptive activity in the abdominal constriction test.
Thus, in the present study, it was found that compound 2d was the most active molecule in all antinociceptive tests. Therefore, this compound was thought to produce both central and peripheral antinociception, while 2a induced only a central antinociceptive effect. Overall, our results confirm that these compounds have a generally peripheral antinociceptive effect.
The compound 2d bears a fluoro group on one of the aryl residues. The other active molecules 2g and 2j bear a methyl and a methoxy on each of the other aryl residues while 2a is nonsubstituted. Although it is known that the mentioned substituents are important for the antinociceptive activity of 1,2-diarylheterocyclic compoundsCitation1, it may not be possible to put forward an idea about the contribution of the substituent to the activity.
Anticancer activity
The compounds selected by NCI and their preliminary anticancer test results as growth percent values obtained against NSCLC (non-small cell lung cancer), BC (breast cancer), and CNSC (central nervous system cancer) cells are given in . These cells were NCI-H460, MCF7, and SF-268, respectively. Compound 2i showed remarkable inhibition values for the cells NCI-H460 and MCF7, but the other compounds were found to be inactive. Compound 2i was accepted for a further screening test. In this step, the selected compound was evaluated in vitro against 66 human tumor cell lines derived from nine neoplastic diseases. The detailed test results are given in .
Table 3. Anticancer activity of some compounds as growth percent against selected cell lines.
Table 4. Mean log10 GI50 values of compound 2i and standard compounds.
According to the test method, it is stated that compounds having growth percent values greater than −4 are considered as inactive. It can be seen that for compound 2i, log10 GI50 (logarithm of concentration that causes 50% growth inhibition) values are smaller than −4. Therefore, we may conclude that the compound provides a notable activity level. Melphalan and cisplatin (cis-diaminodichloroplatinum), two commonly used chemotherapeutic agents, were used as standard compounds. When the mean-graph midpoint (MG-MID) values of the compounds melphalan and cisplatin, i.e. −5.09 and −6.20 respectively, are considered, it is observed that compound 2i provides an acceptable activity level (MG-MID −4.39).
Acknowledgements
The authors present their thanks to NCI (USA) and Anadolu University BIBAM (Turkey) AAQfor Anticancer test results and NMR spectral data respectively.
Declaration of interest: The authors report no conflicts of interest.
References
- Pariet M, Ryn JV. COX-2 Inhibitors. Berlin: Birkhauser Verlag, 2004:15–40, 227–43.
- Talley JJ. Selective inhibitors of cyclooxygenase-2. Prog Med Chem 1999;36: 201–34.
- Leval X, Delarge J, Somers F, Tullio P, Henrotin Y, Pirotte B, et al. Recent advances in inducible cyclooxygenase (COX-2) inhibition. Curr Med Chem 2000;7:1041–62.
- Dannhardt G, Kiefer W. Cyclooxygenase inhibitors—current status and future prospects. Eur J Med Chem 2001;36:109–26.
- Penning TD, Talley JJ, Bertenshaw SR, Carter JS, Collins PW, Docter S, et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-5-(4-metylphenyl)-3-(trifluorometyl)-1h-pyrazol-1-yl-benzenesulfonamide (sc-58635, celecoxib). J Med Chem 1997;40:1347–65.
- Khanna IK, Weier RM, Yu Y, Collins PW, Miyashiro JM, Koboldt CM, et al. 1,2-Diarylpyrroles as potent and selective inhibitors of cyclooxygenase-2. J Med Chem 1997;40:1619–33.
- Talley JJ, Brown DL, Carter JS, Graneto MJ, Koboldt CM, Masferrer JL, et al. 4-[5-Methyl-3-phenylisoxazol-4-yl]-benzenesulfonamide, valdecoxib: a potent and selective inhibitor of COX-2. J Med Chem 2000;43:775–7.
- Habeeb AG, Rao PNP, Knaus EE. Design and syntheses of diarylisoxazoles: novel inhibitors of cyclooxygenase-2 (COX-2) with analgesic-antiinflammatory activity. Drug Dev Res 2000;51:273–86.
- Almansa C, Arriba AF, Cavalcanti FL, Gomez LA, Miralles A, Merios M, et al. Synthesis and SAR of a new series of COX-2-selective inhibitors: pyrazolo[1,5-a]pyrimidines. J Med Chem 2001;44:350–61.
- Hashimoto H, Imamura K, Haruta J, Wakitani K. 4-(4-Cycloalkyl/aryl-oxazol-5-yl)benzenesulfonamides as selective by introduction of a fluorine atom and identification of a potent, highly selective, an orally active COX-2 inhibitor jte-522. J Med Chem 2002;45:1511–17.
- Laufer SA, Wagner GK. From imidazoles to pyrimidines: new inhibitors of cytokine release. J Med Chem 2002;45:2733–40.
- Liu H, Huang X, Shen J, Luo X, Li M, Xiong B, et al. Inhibitory mode of 1,5-diarylpyrazole derivatives against cyclooxygenase-2 and cyclooxygenase-1: molecular docking and 3D QSAR analyses. J Med Chem 2002;45:4816–27.
- Almansa C, Alfon J, Arriba A, Cavalcanti FL, Escamilla I, Gomez LA, et al. Synthesis and structure-activity relationship of a new series of COX-2 selective inhibitors: 1,5-diarylimidazoles. J Med Chem 2003;46:3463–75.
- Rao PNP, Amini M, Li H, Habeeb AG, Knaus EE. Design, synthesis, and biological evaluation of 6-substituted-3-(4-methanesulfonylphenyl)-4-phenylpyran-2-ones: a novel class of diarylheterocyclic selective cyclooxygenase-2-inhibitors. J Med Chem 2003;46:4872–82.
- Hu W, Guo Z, Chu F, Bai A, Yi X, Cheng G, et al. Synthesis and biological evaluation of substituted 2-sulfonyl-phenyl-3-phenyl-indoles: a new series of selective COX-2 inhibitors. Bioorg Med Chem 2003;11:1153–60.
- Singh SK, Vobbalareddy S, Shivaramakrishna S, Krishnamraju A, Rajjak SA, Casturi SR, et al. Methanesulfonamide group at position-4 of the c-5-phenyl ring of 1,5-diarylpyrazole affords as a potent class of cyclooxygenase-2 (COX-2) inhibitors. Bioorg Med Chem Lett 2004;14:1683–8.
- Tuyen TN, Sin K, Kim HP, Park H. Synthesis and antiinflammatory activity of 1,5-diarylimidazoles. Arch Pharm Res 2005;28:1013–18.
- Mozziconacci J, Arnoult E, Bernard P, Do QT, Marot C, Morin-Allory L. Optimization and validation of a docking-scoring protocol; application to virtual screening for COX-2 inhibitors. J Med Chem 2005;48:1055–68.
- Laufer SA, Zimmermann W, Ruff KJ. Tetrasubstituted imidazole inhibitors of cytokine release: probing substituents in the n-1 position. J Med Chem 2004;47:6311–25.
- Navidpour L, Shadnia H, Shafaroodi H, Amini M, Dehpour AR, Shafiee A. Design, synthesis and biological evaluation of substituted 2-alkylthio-1,5-diarylimidazoles as selective COX-2 inhibitors. Bioorg Med Chem 2007;15:1976–82.
- Borne RF. Nonsteroidal antiinflammatory drugs. In: Foye WO, Lemke TL, Williams DA, eds. Principles of Medicinal Chemistry, 4th ed. Philadelphia, PA: Williams & Wilkins, 1995:535–80.
- Hida T, Kozaki K, Muramatsu H, Masuda A, Shimizu S, Mitsudomi T, et al. Cyclooxygenase-2 inhibitor induces apoptosis and enhances cytotoxicity of various anticancer agents in non-small cell lung cancer cell lines. Clin Cancer Res 2000;6:2006–11.
- Petersen C, Baumann M, Petersen S. New targets for the modula- tion of radiation response-selective inhibition of the enzyme cyclo- oxygenase 2. Curr Med Chem Anticancer Agents 2003; 3:354–9.
- Gust R, Busch S, Keilitz R, Schmidt K, Rauch M. Investigations on the influence of halide substituents on the estrogen receptor interaction of 2,4,5-tris(4-hydroxyphenyl)imidazoles. Arch Pharm Pharm Med Chem 2003;336:456–65.
- Ye F, Wu J, Dunn T, Yi J, Tong X, Zhang D. Inhibition of clclooxygenase-2 activity in head and neck cancer cells by genistein. Cancer Lett 2004;211:39–46.
- Johnsen JI, Lindskog M, Ponthan F, Pettersen I, Elfman L, Orrego A. Cyclooxygenase-2 is expressed in neuroblastoma, and nonsteroidal anti-inflammatory drugs induce apoptosis and inhibit tumor growth in vivo. Cancer Res 2004;64:7210–15.
- Kaufmann HP, Huang S, Buckmann HJ. über antipyril-ketone. Berichte 1942;75B:1214.
- Korohoda MJ, Bojarska AB. Methylation of 4-imidazoline-2-thiones. J Prakt Chem 1991;333:355–60.
- Biancchi C, Franceschini J. Experimental observations on Haffners method for testing analgesic drugs. Br J Pharmacol 1954;9:280–4.
- Eddy NB, Leimbach D. Synthetic analgesic (II). Dithienylbutenyl- and dithienylbutylamines. J Pharmacol Exp Ther 1953;107:385–93.
- Noble F, Smadja C, Roques BP. Role of endogenous cholecystokinin in the facilitation of mu-mediated antinociception by delta opioid agonists. J Pharmacol Exp Ther 1994;271:1127–34.
- Bastos GNT, Santos ARS, Ferreira VMM, Costa AMR, Bispo CI, Silveira AJA, et al. Antinociceptive effect of aqueous extract obtained from roots of physalis angulata L. on mice. J Ethnopharmacol 2006;103:241–5.
- Koster R, Anderson M, Beer EJ. Acetic acid for analgesic screening. Fed Proc 1959;18:412.
- Boyd MR. Status of the NCI preclinical antitumor drug discovery screen. Princ Pract Oncol 1989;3:2–11.
- Gürsoy A, Demirayak S, Capan G, Erol K, Vural K. Synthesis and preliminary evaluation of new 5-pyrazolinone derivatives as analgesic agents. Eur J Med Chem 2000;35:359–64.