Abstract
The inhibition of recombinant mouse acetylcholinesterase (rMAChE) and electric eel acetylcholinesterase (EEAChE) by seven, structurally different chromophore-based (dansyl, pyrene, dabsyl, diethylamino- and methoxycoumarin, Lissamine rhodamine B, and Texas Red) propargyl carboxamides or sulfonamides was studied. Diethylaminocoumarin, Lissamine, and Texas Red amides inhibited rMAChE with IC50 values of 1.00 μM, 0.05 μM, and 0.70 μM, respectively. Lissamine and Texas Red amides inhibited EEAChE with IC50 values of 3.57 and 10.4 μM, respectively. The other chromophore amides did not inhibit either AChE. The surprising inhibitory potency of Lissamine was examined in further detail against EEAChE and revealed a mixed-type inhibition with Ki = 11.7 μM (competitive) and Ki′ = 24.9 μM (noncompetitive), suggesting that Lissamine binds to free enzyme and enzyme–substrate complex.
| Abbreviations: | ||
| AChE, | = | acetylcholinesterase; |
| rMAChE, | = | recombinant mouse brain acetylcholinesterase. |
Introduction
Acetylcholinesterase (AChE) activity is essential to neuromuscular and brain function as the principal enzyme responsible for the breakdown of the neurotransmitter, acetylcholine (ACh). Thus, the inhibition of AChE causes a corresponding increase in ACh concentration. When the inhibitor is a carbamate or organophosphate, excitotoxicity occurs through the prolonged innervation of cholinergic receptors due to increased ACh concentration. Using more beneficial AChE inhibitors, for example tacrine, an increase in ACh could potentially delay the onset of Alzheimer’s disease (AD) via this same prolonged, yet controlled receptor activationCitation1.
The structure of AChE is highlighted by a catalytic active site (CAS), a peripheral active site (PAS), and a narrow, yet deep (20 å) gorge that connects the CAS to the PAS. The CAS is located near the bottom of the gorge, whereas the PAS is located near the enzyme surface (substrate entrance) and has been implicated in the formation of amyloid-β fibrilsCitation2. Various AChE inhibitors to the CAS, the PAS, or both have been developed for ADCitation3–5, among which inhibitors linked with fluorophores have been used to probe the mechanism of actionCitation6–8. Most of these studies are based on the assumption that the attached fluorophores play no significant role in AChE inhibition. Recent studies by Kucukkilinc and co-workers, however, showed that certain fluorophores can inactivate AchECitation9–11. To better understand the mechanism of AChE inhibition by fluorophores, seven common structures were selected with and without cation-containing groups and examined against recombinant mouse (rMAChE) and electric eel acetylcholinesterase (EEAChE). The reactive group of each fluorophore (RCO2H or RSO3Cl) was converted to the propargyl amide to provide a sterically benign moiety that also could be used in future studies as a potential coupling partner for click chemistryCitation12,Citation13. The inhibition data will be used to design more efficient AChE probe structures, design imaging agents for AChE-expressing cells, and in FRET (fluorescence energy transfer) experiments to elucidate amyloid formation.
Materials and methods
Chemicals and enzymes
Acetylthiocholine iodide (ATCh-I), 5,5′-dithio(2-nitrobenzoic acid) (DTNB), propargylamine, and EEAChE type V-S were purchased from Sigma-Aldrich (Milwaukee, WI). The seven amine reactive fluorophores (dansyl, pyrene, dabsyl, diethylaminocoumarin, methoxycoumarin, Lissamine rhodamine B, and Texas Red) were obtained from Invitrogen Inc. (Carlsbad, CA). Crude EEAChE was diluted to a 1 mg/mL stock solution in 0.1 M phosphate buffer pH 7.6 (PBS 7.6) at 0–4°C. Soluble recombinant mouse AChE was expressed in HEK 293 cells and purified by customized affinity chromatography as describedCitation14. Stock solutions of rMAChE (1 mg/mL) were prepared in 0.1 M PBS 7.6 and stored at 0–4°C prior to use.
Preparation of the fluorophore amides
The compounds were synthesized using conventional methods. The two coumarin carboxylic acids were activated with a coupling reagent, DIPC (1,3-diisopropyl-carbodiimide) and condensed with propargyl amine to form the coumarin carboxamides 4 and 5. The remaining five fluorophores purchased as the sulfonyl chlorides were reacted with propargyl amine in dry dichloromethane to form the corresponding propargyl sulfonamides 1–3, 6, and 7 as reported by Bolletta et al.Citation15,Citation16. All the fluorophore amides 1–7 () were purified using flash chromatography with mixtures CHCl3/MeOH (95/5), chloroform, or ethyl acetate. The structures of the amides 1–7 were confirmed by nuclear magnetic resonance (NMR) and mass spectroscopy (MS), and prepared in acetonitrile (ACN) as stock solutions (10 mM).
Figure 1. The chemical structures of the fluorophore amides.
5-(Dimethylamino)-N-(-2-propynyl)-1-naphthalenesulfonamide (1) (dansyl propargyl amide)
1H NMR (400 MHz, CDCl3): δ (ppm) 8.54 (d, J = 7.0 Hz, 1H), 8.25 (t, J = 7.0 Hz, 2H), 7.53 (m, 2H), 7.17 (d, J = 7.0 Hz, 1H), 5.01 (brs, 1H, N-H), 3.75 (s, 2 H), 2.87 (s, 6H), 1.89 (s, 1H); 13C NMR (100 MHz, CDCl3): δ (ppm) 152.1, 134.1, 131.0, 130.2, 129.9, 128.8, 123.4, 118.8, 115.5, 77.6, 72.9, 45.7. ESI-MS: calc. for C15H17N2O2S+ (M + H), 289.10; found 289.11.
1-N-(-2-Propynyl)-pyrenesulfonamide (2) (pyrene propargyl amide)
1H NMR (400 MHz, CDCl3): δ (ppm) 8.91 (d, J = 8.0 Hz, 1H), 8.71 (d, J = 8.0 Hz, 1H), 8.33 (m, 1H), 8.24 (d, J = 7.8 Hz, 1H), 8.22 (d, J = 7.8 Hz, 1H), 8.12 (m, 4H), 4.59 (brs, 1H, N-H), 3.82 (d, J = 3.2 Hz, 2H), 1.2 (s, 1H); 13C NMR (100 MHz, CDCl3): δ (ppm) 134.3, 131.2, 130.6, 130.1, 130.0 (2), 129.6, 127.8, 127.4, 127.3, 127.1 (2), 124.1, 123.1, 77.6, 72.1, 41.8. ESI-MS: calc. for C19H14NO2S+ (M + H); 320.07, found 320.06.
4-(4-Dimethylamino-phenylazo)-N-prop-2-ynyl benzenosulfonamide (3) (dabsyl propargyl amide)
1H NMR (400 MHz, CDCl3): δ (ppm) 7.98–788 (m, 6H), 6.80 (d, J = 9.2 Hz, 2H), 4.67 (brs, 1H, NH), 3.87 (d, J = 2.4 Hz, 2H), 3.15 (s, 6H), 2.10 (s, 1H); 13C NMR (100 MHz, CDCl3): δ (ppm) 156.2, 153.8, 143.8, 143.2, 128.4, 126.6, 123.1, 111.8, 77.7, 73.7, 40.5, 33.1. ESI-MS: calc. for C17H19N4O2S+ (M + H), 343.12, found 343.13.
2H-1-Benzopyran-7-N-diethyl-2-oxo-3-propynylcarboxylamide (4) (diethylcoumarin propargyl amide)
1H NMR (400 MHz, CDCl3): δ (ppm) 9.02 (brs, 1H, N-H), 8.69 (s, 1H), 7.43 (d, J = 8.8 Hz, 1H), 6.65 (dd, J = 8.8 Hz, 2.2 Hz, 1H), 6.49 (d, 1H, J = 2.4 Hz), 4.22 (m, 2 H), 3.45 (q, J = 6.4 Hz, 4H), 2.27 (s, 1H), 1.12 (t, J = 6.4 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ (ppm) 163.2, 162.8, 152.9, 148.6, 136.7, 131.4, 110.2, 112.7, 108.6, 96.7, 79.9, 71.4, 45.3, 45.1, 12.6. ESI-MS: calc. for C17H19N2O3+ (M + H), 299.14, found 299.15.
2H-1-Benzopyran-7-methoxy-2-oxo-3-N-propynylcarboxylamide (5) (methoxycoumarin propargyl amide)
1H NMR (400 MHz, CDCl3): δ (ppm) 8.95 (brs, 1 H, N-H), 8.83 (s, 1H), 7.56 (d, J = 8.8 Hz, 1H), 6.94 (d, J = 8.8 Hz, 1H), 6.86 (s, 1H), 4.23 (m, 2H), 3.90 (s, 3H), 2.24 (s, 1H); 13C NMR (100 MHz, CDCl3): δ (ppm) 165.3, 161.9, 148.9, 133.1, 131.3, 130.0, 114.4, 112.5, 106.7, 100.6, 79.6, 71.7, 56.3, 42.9. ESI-MS: calc. for C14H12NO4+ (M+H), 258.08, found 258.08
Xanthylium, 9-[-2-sulfophenyl]-3,6-bis(diethylamino)-4-sulfonamide (6) (Lissamine propargyl amide)
1H NMR (400 MHz, CD3OD): δ (ppm) 8.67 (s, 1H), 8.14 (d, J = 6.8 Hz, 1H), 7.52 (d, J = 8.0 Hz, 1H), 7.20 (d, J = 8.8 Hz, 1H), 7.17 (s, 2H), 7.08 (s, 1H), 7.05 (d, J = 8.0 Hz, 1H), 3.93 (s, 2H), 3.75 (m, 8H), 2.62 (s, 1H), 1.31(m, 12H); 13C NMR (100 MHz, CDCl3): δ (ppm) 167.6, 153.9, 153.7, 153.5, 149.6, 149.1, 134.6, 132.8, 130.6, 129.3, 129.2, 128.2, 124.8, 124.3, 123.9, 123.2, 108.2, 105.3, 97.9, 78.5, 70.2, 44.7, 44.6, 28.7. ESI-MS: calc. for C30H34N3O6S2+ (M + H), 596.19, found 596.25.
9-[2(or 4)-[[[-[-N-Propynylsulfonamide]-4(or 2)-sulfophenyl]-2,3,6,7,12,13,16,17–octahydro-1H, 5H, 11H, 15H-xantheno[2,3,4-ij:5,6,7i′j′]-diquinolizium-18-inner salt (7) (Texas-red propargyl amide)
1H NMR (400 MHz, CDCl3): δ (ppm) 9.07 (s, 1H), 8.56 (d, J = 1.6 Hz, 1H), 7.50 (s, 1H), 6.64 (s, 2H), 5.2 (brs, 1H, NH), 3.52 (m, 2H), 3.48 (m, 4H), 3.02 (m, 1H), 2.72 (m, 1H), 2.10 (m, 1H), 1.96 (m, 1H), 1.58 (m, 8H), 1.28 (s, 4H), 0.88 (t, J = 6.8 Hz, 4H). ESI-MS: calc. for C34H34N3O6S2+ (M + H), 644.19, found 644.20.
AChE assays
The inactivation of AChEs was determined using a colorimetric assayCitation17. Carrier solvents (ACN, methanol, ethanol, or acetone) showed that 1% (v/v) organic solvent caused a significant decrease (>5%) in rMAChE enzyme activity. ACN was selected as a carrier because it showed no effect on enzyme activity at 0.5% (v/v). All the fluorophore amides showed interference at 412 nm at ≥100 μM, and therefore inhibition studies were conducted at ≤100 μM. The inhibition of rMAChE and EEAChE by compounds 1–7 was determined as follows. DTNB solution and a solution of AChE yielding a 0.1 Abs unit/min rate in PBS (2.80 mL; pH 7.6) were placed in a cuvette at 20°C. To this solution was added either: (a) 10 μL of ACN as the control, or (b) 10 μL of ligand solution (10 mM in ACN). After 6 min incubation, the remaining enzyme activity was determined by adding 20 μL aliquots of the ATCh-I solutions and the hydrolysis rate was monitored at 412 nm over a period of 10 min (15 s intervals). The final concentrations of the reactants during enzyme assay were: 0.33 mM DTNB, 0.59 mM ATCh-I, 0.58 mM NaHCO3, and 0.05 mM inhibitor. IC50 values were determined using the same assay solutions as above, except five different concentrations of the inhibitors (10 nM–50 μM) were used, and the remaining enzyme activities were recorded at a set time point. When needed, the inhibitor concentration range was adjusted to cover the AChE inactivation from 20% to 80% to provide valid IC50 estimations. The data were analyzed by Kaleidagraph 3.6 (Synergy Software, Reading, PA), and the IC50 value for each compound at 6 min incubation was determined from the inhibition curve. All experiments were conducted in triplicate.
To further delineate the inhibitory mechanism, Lissamine propargyl amide 6 was assayed against EEAChE at five substrate concentrations (ATCh-I, duplicate experiments). The substrate concentrations (0.1–1.5 mM, final concentrations) were applied after incubation with the inhibitor (0–0.025 mM, final concentrations) for 10 min and the resultant enzyme activity was recorded. Lineweaver–Burk analysis in the presence of different substrate concentrations as a function of inhibitor concentration was conducted.
Results and discussion
Diethylamino- and methoxycoumarin were converted to the corresponding carboxamides 4 and 5 in >80% yield following chromatography. The remaining fluorophores purchased as the sulfonyl chloride were reacted directly with propargyl amine to afford sulfonamides 1, 2, 3, 6, and 7 in >95% yield following chromatography.
The fluorophore propargyl amides 1–7 (50 μM) were tested as inhibitors of rMAChE and EEAChE. Lissamine and Texas Red inhibited both AChEs, and diethylaminocoumarin showed some inhibitory ability toward rMAChE whereas the other fluorophore amides (dansyl, dabsyl, methoxycoumarin, and pyrene) were not inhibitors of either enzyme. Higher concentrations of the ligands were not attempted due to solubility problems and interference with the colorimetric assay. Previous studies showed that EEAChE was competitively inhibited by polycyclic aromatic hydrocarbons, including pyrene with an IC50 of 5.22 μM11. Thus, it was unexpected that the pyrene-linked propargyl amide (2) was inactive as an inhibitor of EEAChE and rMAChE. It is possible that the modest addition of the propargyl sulfonamide group to pyrene adds enough steric interference to reduce access to the gorge or movement through the gorge en route to the CAS.
The concentration-dependent inhibition of rMAChE by diethylaminocoumarin 4, Lissamine 6, and Texas Red 7, afforded IC50 values of 1.00, 0.05, and 0.70 μM, respectively (). Lissamine and Texas Red were also found to inhibit EEAChE with IC50 values of 3.57 and 10.4 μM, respectively (). Diethylaminocoumarin 4 was not an inhibitor of EEAChE. Among the fluorophores tested, 6 showed the strongest anti-AChE activity. From a structural viewpoint, 6 and 7 are also cation-containing fluorophores and can interact with AChE as inhibitors at the PAS, although this cannot be validated from the kinetics. The results are consistent with previously reported anti-AChE activity of positively charged fluorophoresCitation9–11, assuming the fluorophore amides tested in this study bind in the same manner to AChE.
Table 1. Inhibition of rMAChE and EEAChE (IC50 μM; n = 3) by fluorophore amides 4, 6, and 7. Amides 1–3, 5 were inactive at ≥50 μM.
Overall, rMAChE and EEAChE responded similarly to the fluorophore amides with two subtle differences: (a) compounds 4, 6, and 7 showed lower IC50 values for rMAChE than for EEAChE, indicating that rMAChE is more sensitive to fluorophore amide inhibition than EEAChE, and (b) diethylaminocoumarin 4 inactivated rMAChE but not EEAChE, suggesting some structural or mechanistic differences in the binding of this inhibitor. With respect to difference item (b), diethylaminocoumarin is a relatively small inhibitor containing an ester (lactone) group and tertiary amine that could conceivably mimic one or more of the key functional groups of the natural substrate (acetylcholine) and bind at the CAS. However, the ACh–coumarin likeness is limited by the pKa of the coumarin nitrogen that is not a cation at physiologic pH, and the interatomic distance from esteratic oxygen to nitrogen is far longer in the coumarin molecule than in ACh.
To better assess the inhibitor–protein interaction, Lineweaver–Burk analysis of Lissamine 6 was conducted (). The plot reveals that both slopes and intercepts increase with higher inhibitor concentration, demonstrating a mixed-type competitive and noncompetitive inhibition. The competitive inhibition constant of Ki = 11.7 μM was obtained by secondary plot of the slope versus the concentration of 6 (; inset A), and the noncompetitive inhibition constant Ki′ = 24.9 μM was obtained by a secondary plot of the intercept versus the concentration of 6 (; inset B). The data suggest that Lissamine interacts with both the free enzyme and with the enzyme–substrate complex but with different affinities.
Figure 2. Double reciprocal Lineweaver–Burk plot showing the inhibition of EEAChE by Lissamine propargylsulfonamide 6. Inset A: determination of Ki for Lissamine propargylsulfonamide 6 using a plot of the slopes versus inhibitor concentration. y = 0.0446x + 0.523; R2 = 0.996. Inset B: determination of Ki′ for Lissamine propargylsulfonamide 6 using a replot of the intercepts versus inhibitor concentration. y = 108.9x + 2.717; R2 = 0.995.
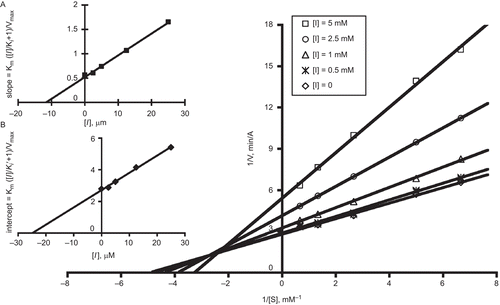
The noncompetitive AChE inactivation by Lissamine indicates that the fluorophore binds to the PAS or some allosteric site other than CAS, while the competitive nature of the AChE inactivation suggests that either there is an overlap between the substrate and inhibitor binding domains or that binding to AChE by the fluorophore blocks the entrance of the gorge, which prevents the substrate from binding to the CAS of AChE. An additional possibility is that the sulfonate anion of Lissamine 6 binds to a region on the enzyme perimeter that alters substrate turnover. Sulfonates such as 2-(N-morpholino)-ethanesulfonic acid (MES) have been found to bind perimeter residues in the AChE crystal structureCitation18–20. However, the involvement of these residues in substrate turnover has not yet been established.
Conclusions
The propargyl amides of two common fluorophores bearing cation groups of 6 and 7 were found to reversibly inactivate rMAChE and EEAChE, whereas dansyl, dabsyl, pyrene, and methoxycoumarin amides were inactive. Diethylaminocoumarin 4 inhibited rMAChE but not EEAChE. Lissamine was found to be a particularly potent inhibitor of rMAChE with an IC50 of 50 nM, and interestingly, the mechanism of inhibition is mixed-type competitive and noncompetitive. Although the cation-containing fluorophores can interact with the PAS, the possibility for interaction with a number of perimeter binding groups that can bind a sulfonate anion cannot be excluded. However, the kinetics suggests the formation of an enzyme–inhibitor complex that prevents the substrate from entering the active site, thereby identifying it as a competitive inhibitor. The results point to the need to carefully select the chemical features of fluorophores in their use as probes, probe constituents, or inhibitor templates in search of new agents to bind the CAS/PAS of AChE. These studies also point to an opportunity to explore Lissamine and cogeners as novel inhibitors of AChE and to utilize their fluorescent properties to decipher the binding loci. We are currently examining this possibility.
Acknowledgments
The research in this study was supported by a grant to one of the authors (C.M.T.) (NIH UO1-ES016102). The authors are grateful for the support of the Core Laboratory for Neuromolecular Production (NIH P30-NS055022) and the Center for Structural and Functional Neuroscience (NIH P20-RR015583).
Declaration of interest: The authors report no conflicts of interest.
Related Research Data
References
- Kihara T, Shimohama S. Alzheimer’s disease and acetylcholine receptors. Acta Neurobiol Exp (Wars) 2004;64:99–105.
- Inestrosa NC, Alvarez A, Calderon F. Acetylcholinesterase is a senile plaque component that promotes assembly of amyloid beta-peptide into Alzheimer’s filaments. Mol Psychiatry 1996;1:359–61.
- McGleenon BM, Dynan KB, Passmore AP. Acetylcholinesterase inhibitors in Alzheimer’s disease. Br J Clin Pharmacol 1999;48:471–80.
- Taylor P. Development of acetylcholinesterase inhibitors in the therapy of Alzheimer’s disease. Neurology 1998;1(Suppl 1):S30–5; discussion S65–7.
- Castro A, Martinez A. Peripheral and dual binding site acetylcholinesterase inhibitors: implications in treatment of Alzheimer’s disease. Mini Rev Med Chem 2001;1:267–72.
- Saltmarsh JR, Boyd AE, Rodriguez OP, Radić Z, Taylor P, Thompson CM. Synthesis of fluorescent probes directed to the active site gorge of acetylcholinesterase. Bioorg Med Chem Lett 2000;10:1523–6.
- Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proc Natl Acad Sci USA 1999;96:14694–9.
- Cravatt BF, Sorensen EJ. Chemical strategies for the global analysis of protein function. Curr Opin Chem Biol 2000;4:663–8.
- Kucukkilinc T, Ozer I. Multi-site inhibition of human plasma cholinesterase by cationic phenoxazine and phenothiazine fluorophores. Arch Biochem Biophys 2007;461:294–8.
- Kucukkilinc T, Ozer I. Inhibition of electric eel acetylcholinesterase by triarylmethane fluorophores. Chem Biol Interact 2008;175:309–11.
- Kang JJ, Fang HW. Polycyclic aromatic hydrocarbons inhibit the activity of acetylcholinesterase purified from electric eel. Biochem Biophys Res Commun 1997;238:367–9.
- Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew Chem Int Ed Engl 2001;40:2004–21.
- Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed Engl 2002;41:2596–9.
- Marchot P, Ravelli RB, Raves ML, Bourne Y, Vellom DC, Kanter J, et al. Soluble monomeric acetylcholinesterase from mouse: expression, purification, and crystallization in complex with fasciculin. Protein Sci 1996;5:672–9.
- Bolletta F, Fabbri D, Lombardo M, Prodi L, Trombini C, Zaccheroni N. Synthesis and photophysical properties of fluorescent derivatives of methylmercury. Organometallics 1996;15:2415–17.
- Lewis WG, Magallon FG, Fokin VV, Finn MG. Discovery and characterization of catalysts for azide-alkyne cycloaddition by fluorescence quenching. J Am Chem Soc 2004;126:9152–3.
- Ellman GL, Courtney KD, Andres V Jr, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95.
- Bartolucci C, Siotto M, Ghidini E, Amari G, Bolzoni PT, Racchi M, et al. Structural determinants of Torpedo Californica acetylcholinesterase inhibition by the novel and orally active carbamate based anti-Alzheimer drug ganstigmine (CHF-2819) J Med Chem 2006;49:5051–8.
- Doucet-Personeni C, Bentley PD, Fletcher RJ, Kinkaid A, Kryger G, Pirard B, et al. A structure-based design approach to the development of novel, reversible AChE inhibitors. J Med Chem 2001;44:3203–15.
- Millard CB, Kryger G, Ordentlich A, Greenblatt HM, Harel M, Raves ML, et al. Crystal structures of aged phosphonylated acetylcholinesterase: nerve agent reaction products at the atomic level. Biochemistry 1999;38:7032–9.